

内标法到底应不应该要求浓度一致
内标法实验的设计思路个人认为有三种类型,第一种是保持上机阶段样品和标曲内标浓度一致,第二种是保持样品和标曲初始加入的内标绝对量一致,样品和标曲内标浓度不必一致,第三种是一种特殊情况,样品上机前加入内标,仅校正基质效应。
从计算便利上来讲,第二种保持内标绝对量一致的设计思路无疑是最好的,计算结果仅需X=c/m,(c为曲线含量,m为样品重量)在工作中也常对一些实验最后定容强行把浓度拉回一致的做法表示不理解,因为前处理的各个阶段的不确定性,样品和标曲的内标响应是不会一致的,既然都需要内标再校正,何必去追求理论上浓度一致?不过最近在进行瘦肉精实验时候,发现了第二种设计思路的弊端,让我认识到保持上机阶段浓度一致是有必要的。
首先看实验部分大概流程:称取5.00g样品于 50 mL 离心管中,加 40 μL 250 μg/L内标溶液,加入 15 mL 乙酸钠-醋酸缓冲液,12000 r/min 离心10 min,分出上层溶液,用盐酸溶液调节至 2.0±0.1(pH 测定仪),进一步 12000 r/min 离心10 min,分出上清液待净化。
净化:依次用水、甲醇、活化平衡固相萃取柱,然后取5mL上述上清液过柱后,用 5mL水、5mL甲醇淋洗除杂,抽干后加入 10 mL 5%氨化乙酸乙酯洗脱收集在 50℃水浴中氮气吹干。先加入 0.1 mL 甲醇超声溶解残留物,再加入 0.9 mL 10%甲醇/水溶液混匀,过 0.22 μm 滤膜后待 LC-MS/MS 分析。
标准曲线的制作:分别吸取 100.0μg/L标准应用液 5 μL、10 μL、20 μL、50 μL 、100 μL,250 μg/L内标混和应用液 40 μL,用 10%甲醇/水溶液定容至 1.0 mL,制备成含量0.5 ng/mL、1.0 ng/mL、2.0 ng/mL、5.0 ng/mL、10 ng/mL 的标准溶液(含 10 ng/mL 的内标溶液)供样品测定用,UPLC-MS/MS 分析后绘制标准曲线。
小结:样品加入内标量为10ng,最终定容到1ml,含内标浓度约为3.3ng/ml(提取液为15ml,取5ml过柱,忽略其他小体积溶液,故浓度为加入量的1/3)。
标准曲线内含内标10ng,浓度 10 ng/mL,典型的样品和标曲内标绝对量一致,上机浓度不一致,计算可不考虑处理过程,可直接用公式:X=c/m。
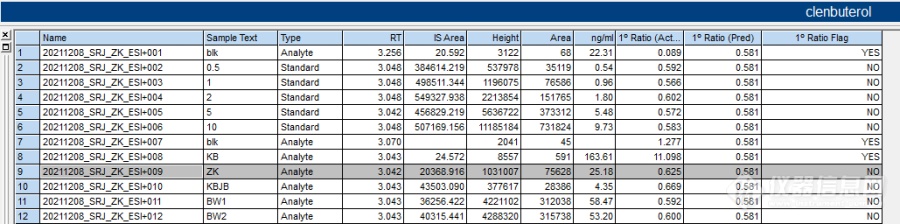
理论上样品和标曲内标响应为1:3,但实际实验却出现一个问题。如图“ IS Area 栏”标准内标响应约为4.7×105,正常处理的样品内内标约为0.4×105(忽略图中个别样品非正常的内标响应)。响应差别达到1:12(理论应为1:3,再加上方法或耗材原因导致的内标损失,雪上加霜),不过待测物响应较好,虽然损失较多,响应还是非常好,积分校正都是正常的。(内标损失严重肯定需要去找原因改进方法但不是这次讨论的重点)。
而从校正结果看(“ng/ml”栏),部分样品含量名义已经超过曲线范围为0.5-10ng/ml,这样给出的结果肯定是不适宜,如果是常规外标法实验,应该重新配制包含样品含量范围的曲线,或者稀释样品。但从样品绝对响应看(“Area栏”),所有样品又均在曲线范围内,只是内标的较大差异,导致出现这个问题,所以这种情况,应该去调整标曲内标的浓度,尽可能匹配样品内的内标浓度。
周智霖
第6楼2023/09/01
??下边是我今天发在论坛的疑问,因为看了大部分论文,他们在预处理前加内标,比如1L水样加10ng内标,最后定容到1ml,但是标曲的绘制的时候,内标浓度为10ng/L,这让我相当疑惑,但是很感谢这篇帖子让我明白了很多
1、内标法需要用不同梯度浓度的标液绘制标准曲线,这个梯度浓度的选择我有个疑问,比如待测样中估算浓度为100ng/L,那我梯度浓度的中间值最好就是100ng/L,但要是我1L水样通过固相萃取,浓缩成了1ml,那我梯度浓度需要改变吗?不改变的话,进色谱的话,待测物的浓度会超过其线性范围
2、和上述一样,内标物的添加量问题,如果前处理过程就加入话,内标会因为前处理,浓度发生变化,这时梯度浓度的标曲中内标物浓度应该为多少?
wsz29
第7楼2023/09/03
1、不管外标法还是内标法,标准曲线范围应该按样品上机液浓度去设计的。2、如果你把处理过程的变化看成损失,曲线加内标量就应该和样品加入的量一致,如果你把处理过程的变化参与计算,曲线加入内标量就应该和样品中内标的真实浓度一致。

































