高效液相色谱法测定甲醛甲酸
崔华宝
摘 要
甲醛装置的液体甲醛,需要分析甲醛中甲酸含量,甲酸是甲醛生产的副产物,监控甲酸指标,能很好的反映装置的反应器运行情况,因此,该数据的准确性对指导装置的良好运行有重要意义。
在装置运行过程中,需要参考实验室分析数据来指导生产和校验在线仪表,在以往分析工作中,发现有时不能快速、准确的提供分析数据。问题出自同一个原因,分析使用的高效液相色谱法谱图异常,目标峰不能被完全的分离,影响积分报告。
为了减小分析偏差、提高时效性的目的。通过优化分析方法,提高分离度和响应值,减少分析时间,保证数据的高效准确。
1背景介绍
1.1. 甲醛中甲酸来源
工艺简介:原料甲醇在并联的甲醇蒸发器中与循环气混合,经与反应气进行热交换后,汽化的甲醇/空气混合气进入两个反应器。反应器类似于管壳式换热器,管中充满催化剂,壳体为联苯基导热油。混合气经过管束时,在催化剂的作用下反应生成甲醛,同时产生大量的反应热,这些反应热,由壳侧的导热油带出。混合气沿管束运行时,温度上升,到达最高点后,温度降低到导热油的沸点附近。管束中的温度最高点称为“热点”,是控制方案中的一个重要参数。
两个反应器的出口气体,在甲醇气化器中冷却后,进入同一吸收工段。吸收工段由两个吸收塔组成,第一吸收塔是填料塔,第二吸收塔是板式塔;反应气体首先进入第一吸收塔,与来自第二吸收塔的液体逆流接触,在塔底生成甲醛产品。甲醛的浓度可以在产品出料管线上自动调配。塔顶气体进入第二吸收塔的底部,经由工艺水吸收后,塔底液体作为第一吸收塔的吸收液,塔顶气体一部分作为循环气返回到反应系统中,其余成为尾气。
甲醛生产的简化主反应式:2CH3OH+O2→2HCHO+2H2O,由于在装置实际运行操作中存在的一些不良因素,例如:催化剂铺装厚度和密度不均匀,造成气流分布不均匀,发生局部过热反应;操作不当,使催化剂床层产生裂缝或塌陷,发生深度氧化反应;原料气夹带液滴,造成催化剂表面结炭;氧温偏低、甲醇原料不合格、塔吸收温度偏高、冷却差、催化剂中毒等。使副反应加剧,简化反应式:2HCHO+O2→2HCOOH,甲醛转甲酸。
由于甲酸的结构特殊,它的一个氢原子和羧基直接相连,也可看做是一个羟基甲醛,因此,甲酸同时具有酸和醛和性质。
健康危害特性:甲醛有特殊的刺激气味,对人眼、鼻等有刺激作用,易溶于水和乙醇,水溶液的浓度最高可达55%,通常是40%,称做甲醛水,俗称福尔马林,是有刺激气味的无色液体,低浓度即可嗅到,人对甲醛的嗅觉阈通常是0.06-0.07mg/m3。长期、低浓度接触甲醛会引起头痛、头晕、乏力、感觉障碍、免疫力降低,并可出现瞌睡、记忆力减退或神经衰弱、精神抑郁;慢性中毒对呼吸系统的危害也是巨大的,长期接触甲醛可引发呼吸功能障碍和肝中毒性病变,表现为肝细胞损伤、肝辐射能异常等。
1.2. 甲醛中甲酸现状分析简介
高效液相色谱技术:高效液相色谱法(HPLC)是一种新型分析与检测技术,具有高效色谱柱、高压输液泵、高灵敏度检测器。广泛应用于食品、药品、农产品、化工产品等分析分离与检验中,具有分析速度快、分析准确度高、高灵敏度、操作简便等优点。配备计算机,数据自动处理,直接绘图和打印分析检测结果,可以对仪器的全部操作进行程序控制,操作简单便捷。高效液相色谱仪是实现液相色谱分析的仪器设备。
甲酸分析原理:甲酸中的羧基中羰基氧和羟基氧孤对电子共轭作用,使其在 205-215nm 处具有紫外吸收,利用甲酸在 210nm 处有紫外吸收,用外标定量。
方法校准记录:
Method 方法名称
|
液体甲醛中甲酸
|
Calibrated by 校准人
|
崔华宝
|
Calibrated on 校准日期
|
20220519
|
Validated to 校准有效期
|
20230518
|
Calibration Type 校准方式
|
外标法
|
Conclusion 校准结论
|
通过
|
试剂记录
|
所需试剂:甲酸钠(AR):纯度≥98.50%,一级水,1+1盐酸
|
一、标准母液配制
|
准确称量0.1504g甲酸钠至100ml容量瓶中,加入1滴(1+1)盐酸溶液,用一级水定容至98.4893g,实际配置的甲酸溶液浓度应为0.103306%(0.1504g*0.6765*100%/98.4893g)。
|
二、标准使用液配制
|
1) 准确称量1.0043g标准母液至100ml容量瓶中,加入1滴(1+1)盐酸溶液,用一级水定容至98.8867g,实际配置的甲酸溶液浓度应为0.001049%(1.0043g*0.103306/98.8867g)。
|
2) 准确称量1.5054g标准母液至100ml容量瓶中,加入1滴(1+1)盐酸溶液,用一级水定容至98.7941g,实际配置的甲酸溶液浓度应为0.001574%(1.5054g*0.103306/98.7941g)。
|
3) 准确称量2.0061g标准母液至100ml容量瓶中,加入1滴(1+1)盐酸溶液,用一级水定容至99.0294g,实际配置的甲酸溶液浓度应为0.002093%(2.0061g*0.103306/99.0294g)。
|
4) 准确称量2.5041g标准母液至100ml容量瓶中,加入1滴(1+1)盐酸溶液,用一级水定容至98.9601g,实际配置的甲酸溶液浓度应为0.002614%(2.5041g*0.103306/98.9601g)。
|
5) 准确称量3.0307g标准母液至100ml容量瓶中,加入1滴(1+1)盐酸溶液,用一级水定容至98.9532g,实际配置的甲酸溶液浓度应为0.003164%(3.0307g*0.103306/98.9532g)。
|
5) 准确称量3.9990g标准母液至100ml容量瓶中,加入1滴(1+1)盐酸溶液,用一级水定容至98.9505g,实际配置的甲酸溶液浓度应为0.004175%(3.9990g*0.103306/98.9505g)。
|
三、标准使用液浓度
|
标准级别
|
甲酸含量%
|
1
|
0.001049
|
2
|
0.001574
|
3
|
0.002093
|
4
|
0.002614
|
5
|
0.003164
|
6
|
0.004175
|
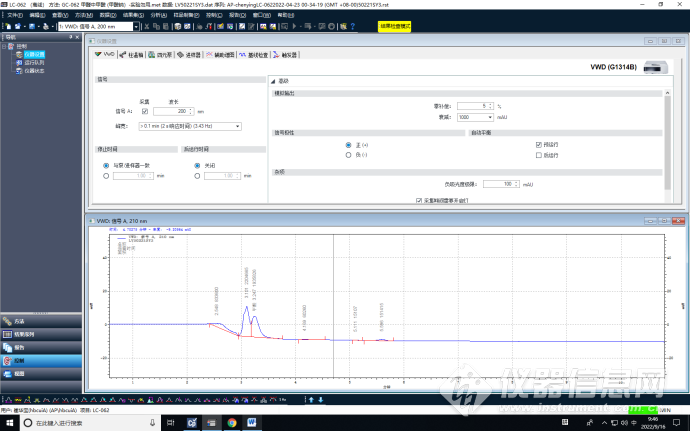
仪器参数截图如下:
VWD
柱温箱
四元泵
辅助谱图
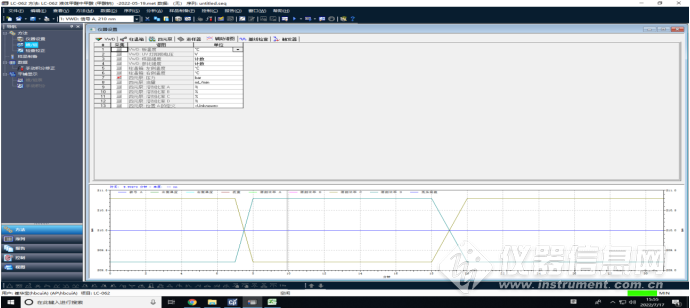
检查校正
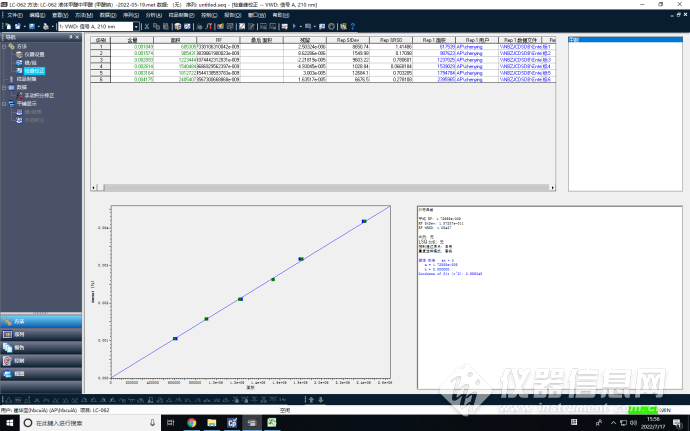
方法检查结果
|
样品描述
|
组分名称
|
理论值%
|
旧校准
结果%
|
新校准
结果%
|
新旧校正结
果相对偏差
|
回收率%
(95~105)
|
甲酸标准
验证
|
甲 酸
|
0.002415
|
0.002423
|
0.002444
|
0.43%
|
101.20
|
方法验证数据截图如下:
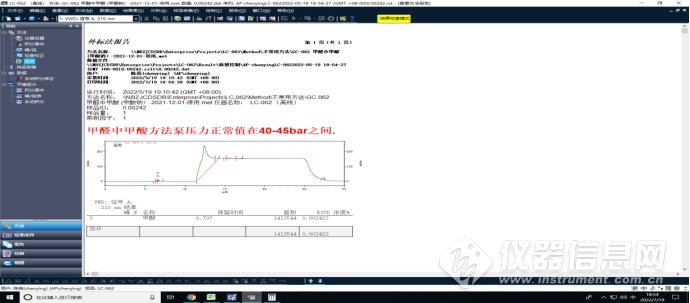
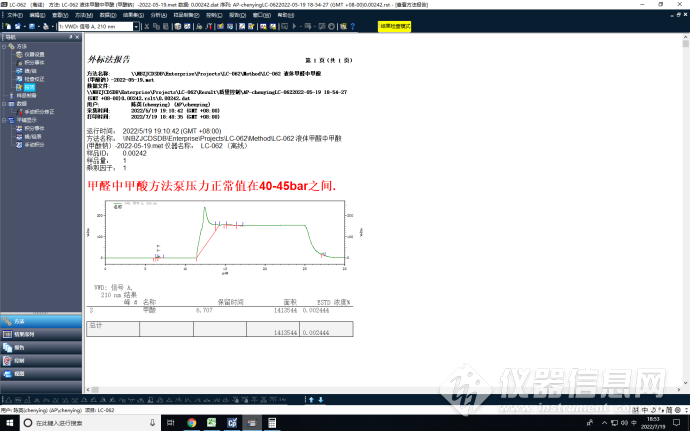
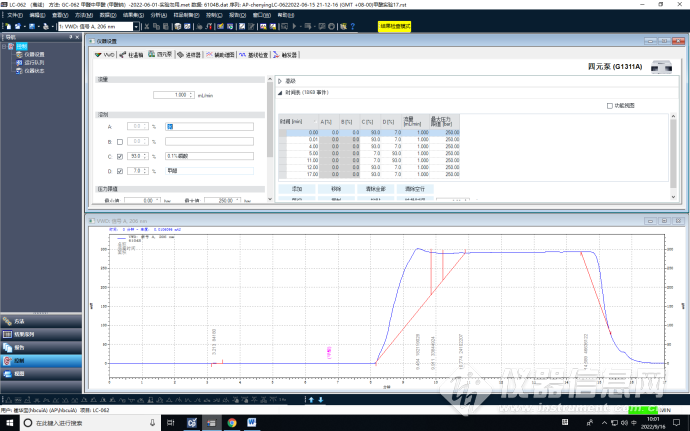
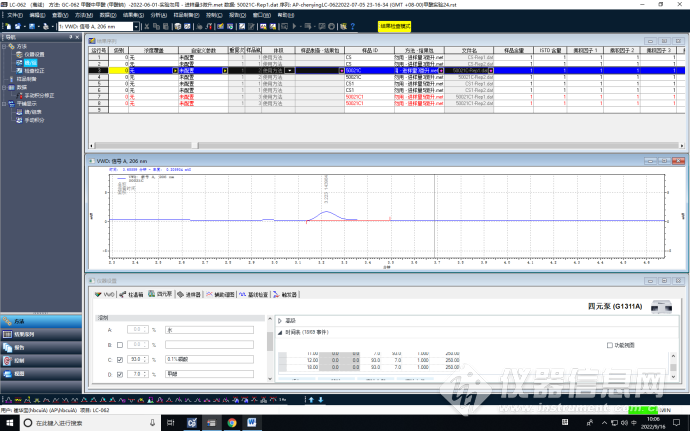
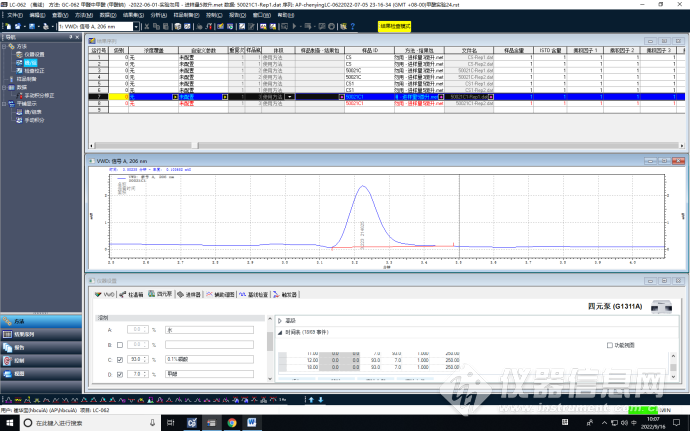
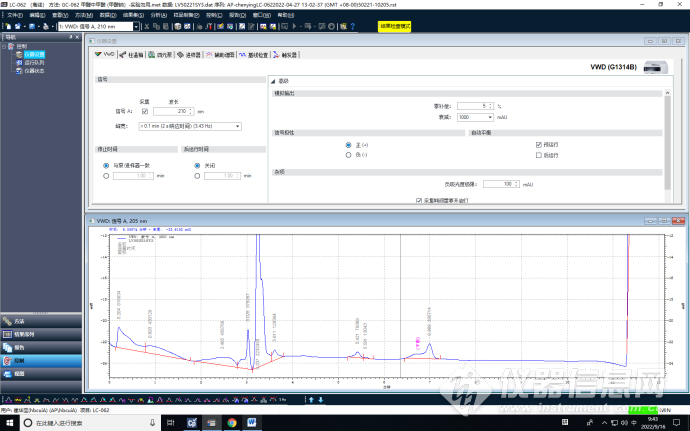
举例实验室样品LV-50021分析谱图及时间:
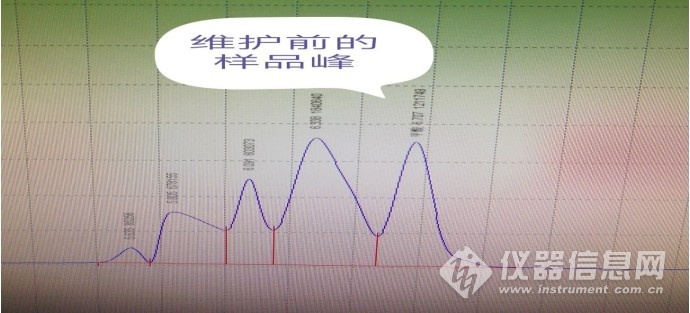
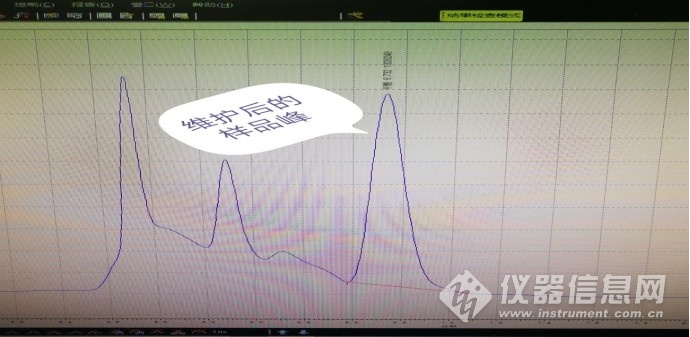
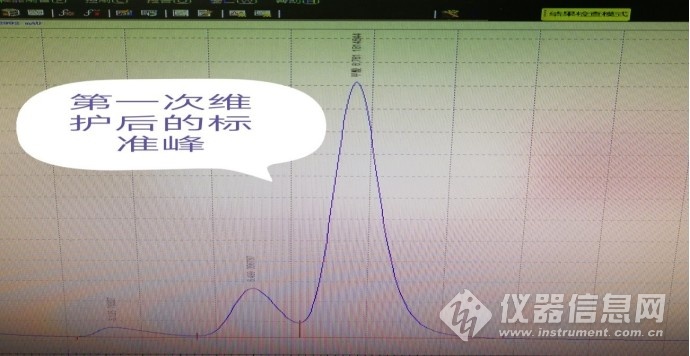
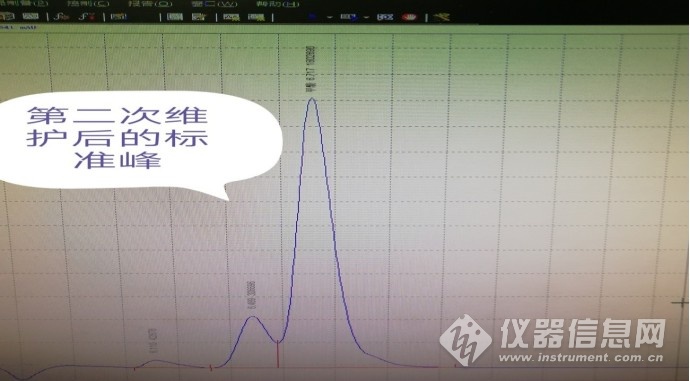
现状:
? 高效液相色谱法,甲酸分析,谱图异常,干扰峰较多,目标峰不能被完全的分离,影响积分报告的可能性高;
? 高效液相色谱法,甲酸分析时间较长;
综合以上现状,该分析方法的质量控制相对受限,数据的时效性受限。
1.3. 优化目标
? 分析方法干扰峰多的原因,找到问题关键点,优化甲酸分析方法。
? 提高目标峰分离度,降低分析出现异常频率,减少分析时长,提高分析效率。
2具体开展工作
2.1原因分析
样品分析的流程图:
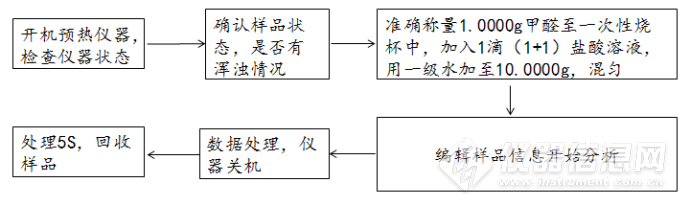
设备本身的稳定性和精度:
安捷伦1260Infinity标准自动进样器的性能指标,在装有100微升标准计量头时有效进样量范围。
1、0.1-100微升,增量为0.1微升带多次抽取升级工具包(需要改动硬件)时最高达1500微升;
2、精度:在5-100微升内,<0.25%RSD,1-5微升以内,<1%,可变体积;
3、样品黏度范围:0.2-50cp。
安捷伦1200柱温箱性能Agilent1200系列柱温箱G1316A/B/C。
1、温度范围,玻耳帖加热和冷却,从低于环境温度10℃直到80℃C(G1316A),高速加热和冷却,具有最大的灵活性和稳定性;
2、最高80℃:流速最高5ml/min G1316A/G1316B,温度稳定性±0.15℃G1316A;
3、加热/冷却时间,从环境温度加热到40℃需5分钟,从40℃冷却到20℃需要10分钟。
从以上高效液相色谱仪性能指标确认,现方法设置的仪器参数,是符合设备本身的稳定性和精度要求的。
针对甲酸分析现状,进行文献查找,通过人机料法环测5M1E法进行原因分析:
1、通过文献查找,确认了高效液相色谱法测定甲酸含量时,缓冲液浓度、pH值、甲醇含量、柱温、流速等配置不同,对色谱分离、溶液抑制解离、改善甲酸的保留和分离的影响,以及流动相对k的影响。
2、缓冲液浓度对容量因子k的影响,随着H3PO4浓度增大,试剂H+的浓度增大,甲酸的k略有增加,随后又下降,但对分离度影响不大,反而酸度太低,会引起硅烷化键合相的降解。流动相pH对k的影响,结果随着流动相pH的增加,甲酸的k下降,这是因为流动相pH增加,甲酸的解离平衡,H+向右移动,离子化程度增加,亲水性增加,结果k下降。由于甲酸为弱酸,易在水系流动相中发生解析,而不被非极性键合相保留;为了改善峰形,减少尾拖尾,可用磷酸缓冲溶液作流动相,以甲醇作有机改性剂,随着流动相中甲醇含量的增加,甲酸的保留时间下降,这是由于甲醇的加入,减弱了甲酸的疏水端与固定相的作用,从而缩短了保留时间。柱温40℃峰形较好,超过40℃甲醇易产生气泡,影响基线稳定。流速对k的影响,流速1.0ml/min为宜,大于1.0ml/min洗脱太快,不利于分离,且柱压升高,不利于柱子的保护和长期使用。
3、在对样品的实际分析应用中,样品组分的复杂性,也对样品分析,带来了干扰因子,进一步,使用Cary 60 紫外-可见分光光度计对样品进行全波长扫描,确认甲酸最佳吸收波长。紫外可见吸收光谱是由于分子中的某些基团吸收了紫外可见辐射光后,发生了电子能级跃迁而产生的吸收光谱。由于各种物质具有各自不同的分子、原子和不同的分子空间结构,其吸收光能量的情况也就不会相同,因此,每种物质就有其特有的、固定的吸收光谱曲线,根据吸收光谱上的某些特征波长处的吸光度的高低判别或测定该物质的含量,这就是分光光度定性和定量分析的基础。Cary 60 紫外-可见分光光度计是一款双光束仪器,配备的强大氙灯每秒可闪烁 80 次,可在3秒内扫描全波长范围(190-1100nm)。氙灯仅在采集到数据时才照射样品,因此可防止敏感样品发生光分解,并降低功耗。高聚焦光束是准确、可重现地测量小样品量的最佳选择。
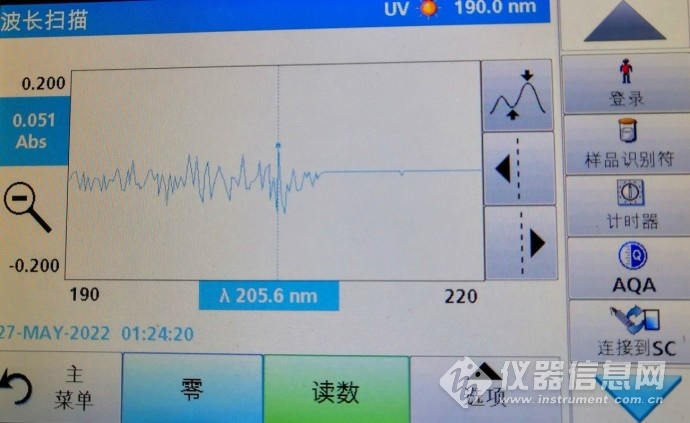
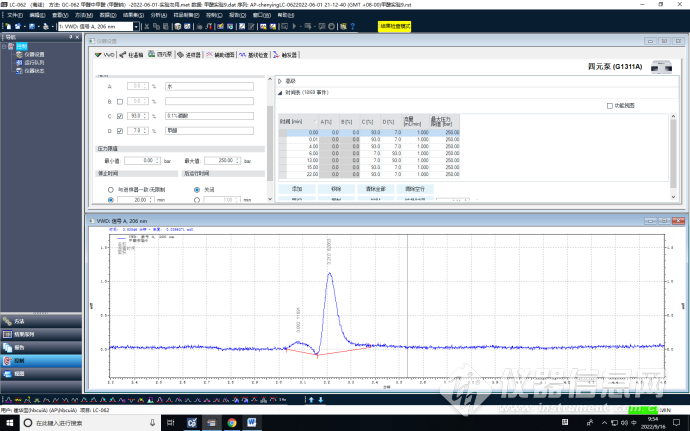
通过对前处理待进样品的波长扫描谱图,可以看到样品的最佳吸收波长为206nm,但最佳吸收波长附近干扰峰也是存在的,现分析方法使用的检测波长为210 nm。
甲酸高效液相法分析谱图,干扰峰较多,经过文献查阅和技术人员交流,可能是样品的进样量30微升较大,造成的。进样量主要由样品性质、色谱柱容量、检测灵敏度和进样系统等决定,进样量过大,保留时间会变化,峰展宽、杂峰多,分离度变差;若组分含量低,溶剂拖尾峰可能掩盖组分峰或难以定量。进样量小,可以克服上述问题,使峰分离良好,分析准确度提高,但保留时间会拖后;若样品组分含量相差较大,微量组分可能难以检出。
4、故最佳色谱分离条件为:柱温40℃,流量1.0mL/min,样品用流动相定容,紫外检测波长206nm,采用CAPCELL PAK-C18色谱柱(4.6mm×250mm,5μm), 甲酸的分离流动相为7%CH30H-0.01mol/L H3PO4 (pH=2.5)时,进行梯度洗脱。
5、分析人员均是经过岗位技能培训,并通过上岗考核合格的,具备相应的分析技能;使用的试剂是具备质量管理资质的厂商提供的分析纯;分析实验室温湿度可控,空气净化度高。依据分析经验,通过谱图的高度一致性,确认人机料环测带来的影响,不是造成分析现状的主要原因。
6、根据原因分析,样品的进样量过大和检测波长参数的设置不合理,可能是导致谱图异常,干扰峰较多,目标峰不能被完全的分离的主要原因。
2.2方法优化与数据收集
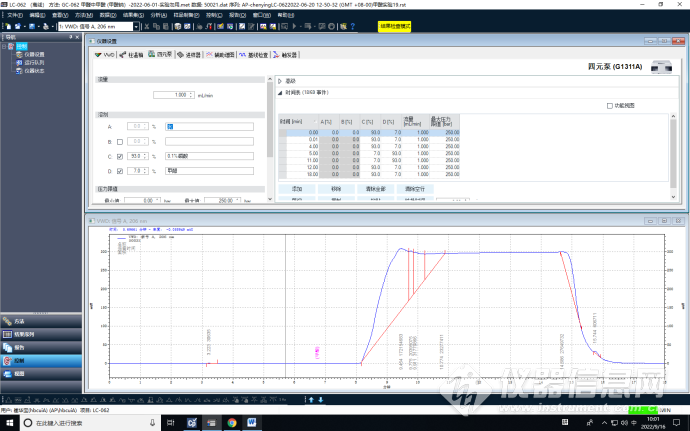
根据上述原因分析和理论最佳色谱分离条件参照,可以先试试减少进样量,以及从流动相入手,通过流动相的比例、流速、柱箱温度,调节分离样品;使用梯度洗脱,调节不同时间的流动相比例,这样既可以达到好的分离效果,又缩短分析时间。
具体实施步骤为:
1、优化甲醛样品前处理的方式,由准确称量1.0000g甲醛至一次性烧杯中,加入1滴(1+1)盐酸溶液,用一级水加至10.0000g,混匀备用;改为准确称量1.0000g甲醛至一次性烧杯中,使用0.1%的磷酸流动相加至10.0000g,使用0.22μm过滤头过滤,混匀后备用。
甲酸钠标准
乙酸钠标准
加入1滴(1+1)盐酸溶液,用一级水加至10.0000g
使用0.1%磷酸稀释至10.0000g
通过甲酸钠、乙酸钠标准运行保留时间的差异和不同前处理方式产生的干扰峰,谱图叠加判断,可以判定,甲酸干扰峰中没有乙酸峰,但含有盐酸带入的干扰峰。
2、优化减少进样量,由30微升进样量,调整为10微升。
进样量1微升
进样量2微升
进样量3微升
进样量5微升
进样量10微升

通过优化进样量可以看到,进样量1微升时,谱图干扰峰最少,峰形较好,利于积分。
3、优化调节紫外检测波长,由210 nm ,调整为最佳吸收波长206nm。
波长200nm
波长205nm
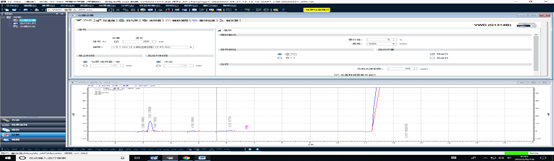
波长206nm
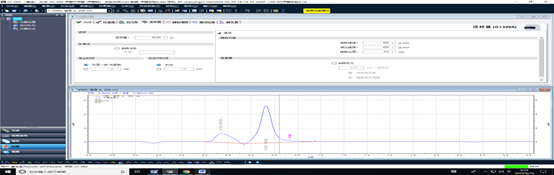
波长210nm
优化调节后紫外检测波长为波长206nm时,具有最好的检测响应,同时减少了干扰峰。
4、优化调整流速,由0.5mL/min流速,调整为1.0mL/min。
流速0.5mL/min
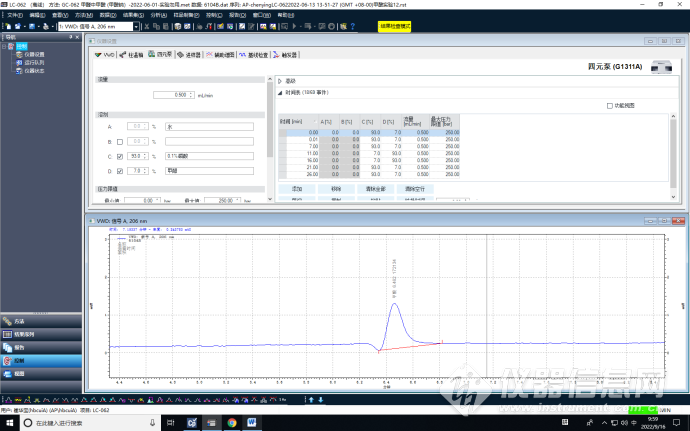
流速1.0mL/min
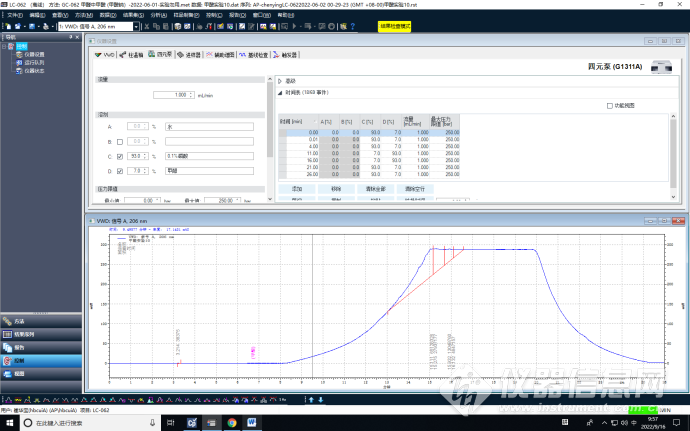
优化调整了流速,增加了柱压,提高了柱效,减少了分析时长。
5、优化增加甲醇比例,由99%0.1%的磷酸+1%甲醇,调整为7%CH30H-0.01mol/LH3PO4 (pH=2.5),进行高压梯度洗脱。
甲醇100%
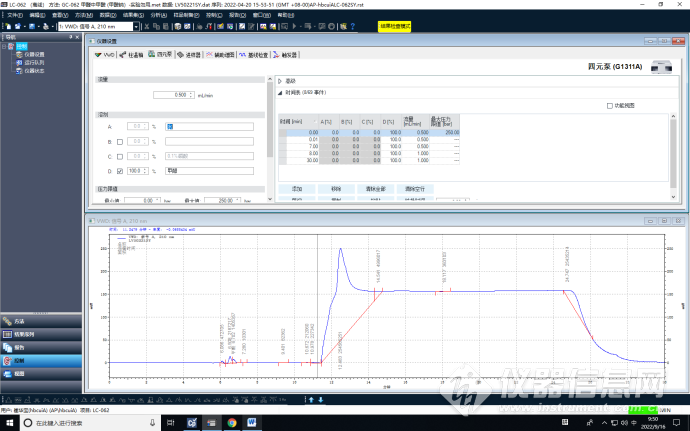
甲醇25%
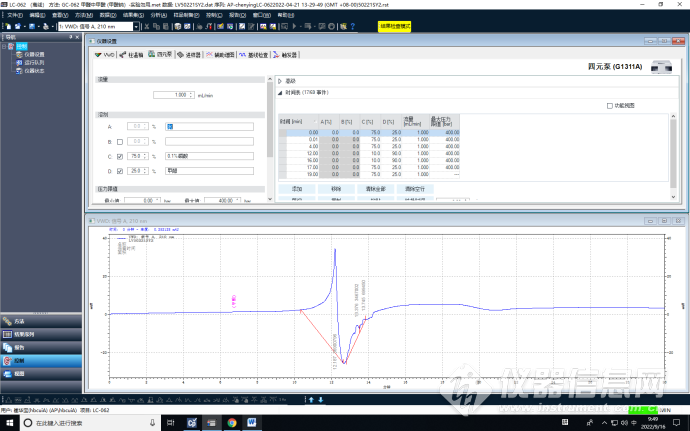
甲醇10%
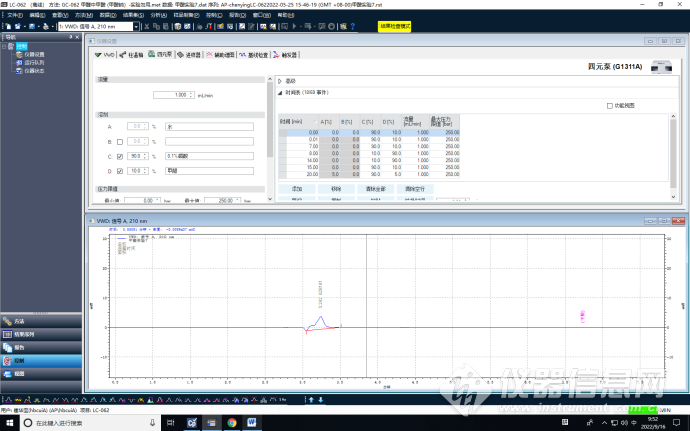
甲醇7%
甲醇5%
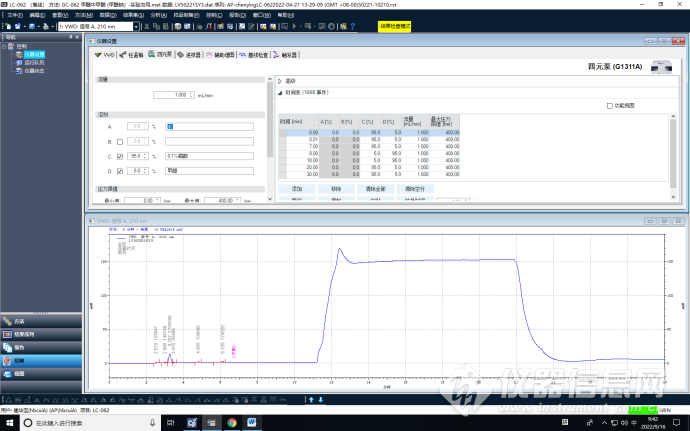
优化增加甲醇比例到7%,保护了色谱柱的同时,降低了保留时间,且峰形较好。
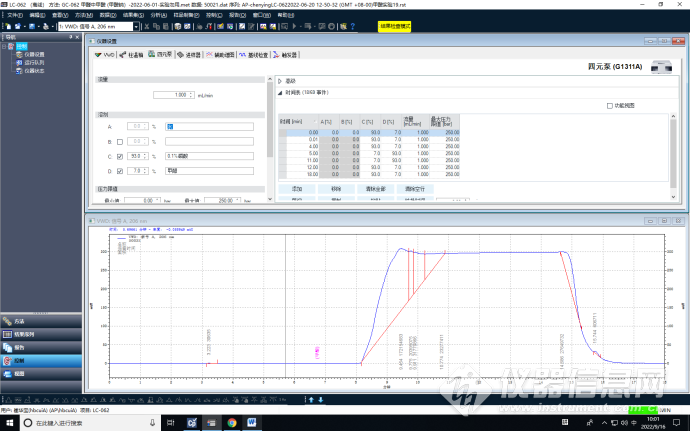
6、优化调节不同时间的流动相比例,由0-7min+20-30min 99%0.1%的磷酸+1%甲醇、8-18 min1%0.1%的磷酸+99%甲醇,调整为0-4min+12-18min93%0.1%的磷酸+7%甲醇、5-11min7%0.1%的磷酸+93%甲醇。
0-17min
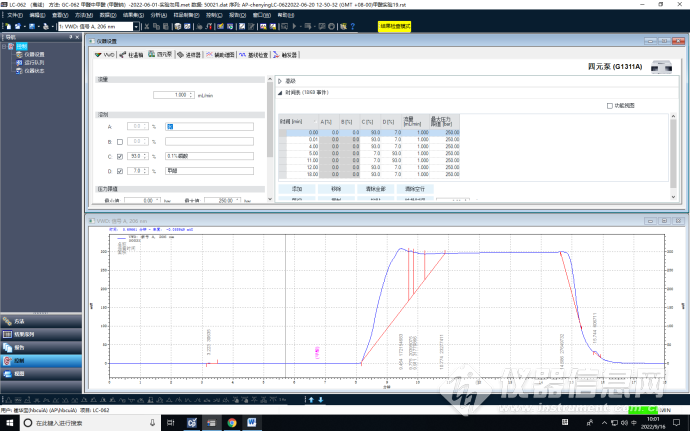
0-26min

优化调节不同时间的流动相比例,稳定柱效的前提下,提高了分离度,降低了分析时长。
7、依据上述实验,得出较为理想的高效液相色谱法的参数设定值:
准确称量1.0000g甲醛至一次性烧杯中,使用0.1%的磷酸流动相加至10.0000g,使用0.22μm过滤头过滤,混匀后备用;进样量10微升,吸收波长206nm,流速1.0mL/min,流动相和梯度为0-4min+12-18min93%0.1%的磷酸+7%甲醇、5-11min7%0.1%的磷酸+93%甲醇。
按照标准方法,配置6个不同浓度的甲酸标准溶液,依次为:0.00104%、0.00155%、0.00207%、0.00258%、0.00307%、0.00409%,进行仪器曲线校正。
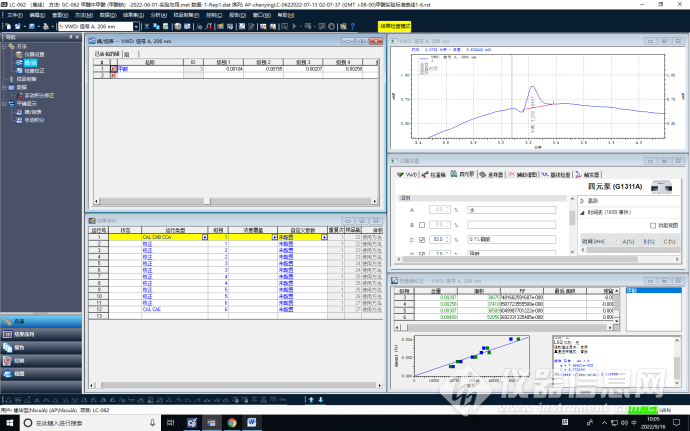
校正后,得到一条完整曲线。
2.3两个方法的相互验证
配置20个新的浓度点,对新老方法进行验证比较:
| 样品新老方法-数据比对 |
编号
| 标准 | 新 | 老 | 差值 | 分析员 |
| 1 | 0.00108 | 0.00110 | 0.00111 | -0.00001 | 1 |
| 2 | 0.00115 | 0.00117 |
0.00116
| 0.00001 | 1 |
| 3 | 0.00120 | 0.00121 | 0.00123 | -0.00002 | 1 |
| 4 | 0.00154 | 0.00157 | 0.00155 | 0.00002 | 1 |
| 5 | 0.00158 | 0.00160 | 0.00162 | -0.00002 | 1 |
| 6 | 0.00160 | 0.00162 | 0.00165 | -0.00003 | 1 |
| 7 | 0.00204 | 0.00207 |
0.00206
| 0.00001 | 1 |
| 8 | 0.00210 | 0.00213 | 0.00215 | -0.00002 | 1 |
| 9 | 0.00216 | 0.00222 | 0.00217 | 0.00005 | 1 |
| 10 | 0.00251 | 0.00253 |
0.00257
| -0.00004 | 1 |
| 11 | 0.00259 | 0.00262 | 0.00263 | -0.00001 | 1 |
| 12 | 0.00262 | 0.00265 | 0.00264 | 0.00001 | 1 |
| 13 | 0.00308 | 0.00311 | 0.00315 | -0.00004 | 1 |
| 14 | 0.00311 | 0.00317 | 0.00312 | 0.00005 | 1 |
| 15 | 0.00325 | 0.00327 | 0.00331 | -0.00004 | 1 |
| 16 | 0.00352 | 0.00356 | 0.00348 | 0.00008 | 1 |
| 17 | 0.00375 | 0.00386 | 0.00380 | 0.00006 | 1 |
| 18 | 0.00380 | 0.00385 | 0.00393 | -0.00008 | 1 |
| 19 | 0.00382 | 0.00395 | 0.00387 | 0.00008 | 1 |
| 20 | 0.00463 | 0.00465 | 0.00471 | -0.00006 | 1 |
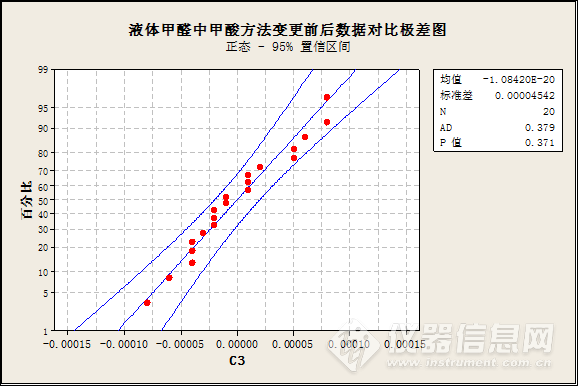
偏差值正态性检验,P值>0.05,成正态分布,进行配对T检验。
配对 T 检验和置信区间:原方法, 新方法
原方法—新方法 的配对 T
平均值
N 平均值 标准差 标准误
原方法 20 0.002596 0.001052 0.000235
新方法 20 0.002596 0.001050 0.000235
差分 20 -0.000000 0.000045 0.000010
平均差的 95% 置信区间: (-0.000021, 0.000021)
平均差 = 0 (与 ≠ 0) 的 T 检验: T 值 = -0.00 P 值 = 1.000
通过上述方法进行验证后,液体甲醛中甲酸原分析方法和新分析方法偏差成正态分布,对两组数据进行配对T检验,P值大于0.05,显示两组数据没有显著差异。可以用新分析方法来进行实验分析。
3结论
综上所述,优化分析前处理方法和仪器参数,可以减少干扰峰,提高目标峰分离度,降低分析出现异常频率,减少分析时长,提高分析效率。选择合适的参数后,分析结果没有显著差异。
4参考资料
【1】高效液相色谱法基本原理与小颗粒色谱填料
【2】万宇 高效色谱法测定甲酸盐中甲酸《饲料博览》2020年第7期
【3】辛梅华.测定雨水中有机酸的方法《分析化学》2003年第1期



































