happy王子矜
第1楼2008/12/15
样品在色谱柱内的情况,我们可以打个比方,大家都看过电影节走红地毯的明星们吧,把色谱柱看成红地毯,然后一群明星(样品)要走过红地毯,地毯旁边会有一大群的记者和影迷,他们就是色谱柱的填料,假设治安状况不够好,明星都需要带着保镖过地毯,保镖我们认为是推着样品前进的流动相,由举办电影节的我们雇佣使用。我们的目的,就是把不同影片的明星分开来,地毯旁的记者影迷(色谱柱填料),负责跟明星握手合影等,这样人气旺的行进速度就会慢,没人搭理的明星们就走的快,在地毯那头,我们的目的达到,明星们分开了。我要开始理论解释了(汗一个先),我们是电影节的组织者,我们要知道那些明星收了出场费但是人没来,所以必须点清明星的个数。在这个过程中,保镖(流动相)是我们雇佣的,有什么问题优先找他们,影迷(色谱柱)是其次可以下手的,明星(样品)我们得罪不起,一般不考虑改变他们来达到目的。就跟踢球踢不过人家,你可以买球员请教练,但是你不能换对手一样;我们对样品没有选择权。给你什么你就得做什么。
合适的检测方法要保证:
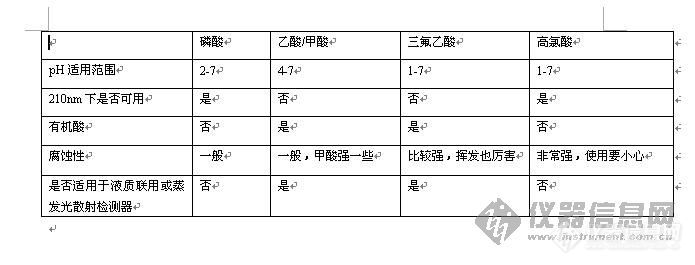
第一,保镖不要对明星太客气,让他们一拥而上过去握手拍照,队形就会拉的很长很散,也许来自同一部影片的明星们都要被分成两队了,这严重影响我们数人头,所以不允许。(流动相的pH和样品的Pka不能接近,否则会出现很难看的峰型,有时甚至出现分叉峰。)
第二,保镖们不要太凶悍,或者明星不要太冷门,这样两边的影迷记者没机会或没兴趣跟明星亲密接触,哗啦啦冲过去了,我们的目的没达到,不行。(选择的色谱柱的固定相要对样品有一定保留,不然样品一下过去,没有任何保留直接出峰,这样的条件是不可行的,因为色谱柱没起到作用,样品里面的东西完全没分离。)
这样的情况下,我们首选考虑换一批不那么凶的保镖(调整流动相的极性,或是降低有机相的比例一般可以改善出峰太早的情况);还有些明星们心急一下子冲过去,这么不给面子的,一定要保镖拦着,保持队形通过(抑制扩散快的样品需要极性强于他的流动相,很多酸性稍强的有机酸,如果水相不加其他东西只是纯水,就会不出峰——因为混在人群中过去了你没看到——或出峰很快,选择比它强的酸是很常见的做法);最后,我们不能怪明星们名气太低,人都是你请的啊,没办法,换一批对他们感冒的粉丝站两边吧。(样品就那样,我们不能改变,如果你使用了你认为合适的流动相体系,样品还是在2分钟或更早就出来,那建议考虑换跟柱子。有些极性特别明显的样品,氰基柱或氨基柱都是不错的选择,或者他们刚好有CN或NH2的基团,这样效果最明显)
第三,跟上面相反,明星太受欢迎,半天移动不了几步,这样我们要调整保镖,拉开旁边太狂热的粉丝们,冲过去。(保留时间太长,出峰太慢的时候,别犹豫,不改变分离的情况下加大有机相比例吧,出峰晚对柱效影响也很大)
第四,某几个明星人气相差很小,影迷对他们的喜欢不相伯仲,于是合伙过地毯——不行(某几个峰不能分离的情况。)
这种情况有以下几种解决方法,大家众所周知的,降低有机相比例可以改变出峰时间,这是首选方法。需要补充一点的是,降低了有机相的比例,只会使所有的峰都后移——我知道有前移的,咱不说那个——但是如果本来分不开的两个峰,前峰的往后跑的多,后峰的往后跑的少,那么调整可能带来相反结果,两个离得更近了,这点要注意。改变缓冲盐的种类或加大缓冲盐的浓度有时也可以改变分离度,一般来说,这对于由峰型不好引起的分离不好作用明显一些。
其次,我们可以换一批更狂热的粉丝团——色谱柱。
狂热的粉丝们不比中庸的C18,看到脸熟都要去握个手,他们喜好明显(对于有苯环,杂环的样品,苯基柱会比C18好,有CN,NH2基团的样品,当然用对应的色谱柱了,C18和C8的差别选择也可以根据这个来判断)。
记住,我不是一上来就推荐你用C18以外的柱子,能用C18这样常用的柱子当然选择它了,不行才选其他的。
这里顺带简要介绍一下色谱柱。有位达人说,C18柱的分离,其实就是样品水溶性差异的分离。在大多数情况下,我认为这个道理成立。C18这么长的一个碳链,极性是非常小的,样品在流动相和色谱柱中间的分配,基本都取决于水溶性差异。但是,现实情况是,在一根C18柱上不能分离的两个峰,换一跟完全分离了,还很漂亮,这跟理论不一样啊。按我理解,这是因为现代色谱柱的特点引起,各个品牌的C18柱都有各自的封尾技术,就是在闲置的Si-OH上面搞,有的是在他们上面连了个CH3——甲基,有的是异丙基,有的干脆把两个Si-OH脱一个水连起来,也有搞包被技术隔离Si-OH的,这就使原本没极性的C18柱带上了C1,C3等等极性相对大很多的基团(好像还有上苯基的,非罗门好像有一款是C1-C6都加的有,还特别宣传过,不过不太确信,忘记了),这样一来,不同C18柱的分离差异相差就有了较大的区别。在某方面来说这是好事,但也会有其他的麻烦,新上的基团会对某些样品有特殊的分离作用,这个我们欢迎,但是新加上去基团的数量会严重影响样品的保留时间。你会看到有的色谱柱宣传他们对流动相比例的改变响应非常敏感,稍微改变比例就会引起分离度的较大差别,有的色谱柱则强调它们各个批次间的差异非常小,你知道原因了吧。对于开发检测条件,前者当然方便,对于长期固定方法检测,后者要好,大家各取所需即可。
最后,前面两种情况都无法改变,或者你觉得换色谱柱很麻烦,那么我们还有大招——让明星们自带粉丝团。这就是离子对试剂。有些样品往往会出现峰型很怪,馒头,山坡等,这是因为保镖和明星们关系不错,限制太少,没有让排成一列横队前进,而是放任他们跑前跑后跟影迷握手(样品的电解平衡点跟流动相很接近,出现了两种形态,离子态和分子态),这跟前面有个情况很相似,正常情况下我们可以用加强保镖力度的方法解决(使流动相的pH和样品的PKa相差2以上),但是遇到大牌明星,你管的太严,人家脸一黑,径直过了红地毯谁也不搭理,你没办法了吧(pH因为不能过7,只能往小了调,改的太低,会影响保留时间)。这个时候,只好请来明星们的铁杆粉丝团,簇拥他们过地毯,这样行进速度慢了下来,对外围的伪粉丝骂退,真粉丝或记者和里面铁杆都是熟人,大家该合影的合影,该打招呼的打招呼,保持了队形,还让不同的明星分离开来。问题解决。(离子对试剂和样品结合,然后色谱柱对其进行保留)这样做的坏处就是清场比较困难,明星都走半天了,铁杆还不愿意散去——你还是打落牙齿咽在肚子里面吧,谁让人家是大牌呢?(离子对试剂使用前,色谱柱需要稳定比较长的时间,运行后的清洗也需要比一般缓冲盐更长的时间,有的色谱柱宣传他们对离子对试剂稳定很快,可见他们是非常职业的记者粉丝团,为我们电影节的组织人员提供了方便。)
上面举例为了说明情况,是按照一般的色谱原理比喻的。其实我们常用的反相色谱,分离模式大多数时候刚好相反,也就是说,两边站的影迷不一定是来拍照合影,而是相当部分都拿着臭鸡蛋准备砸的,这个时候的分离不是看谁的人气高吸引力强出的晚,其实是看谁更不受欢迎谁就跑的更快。其实我觉得道理都一样,换个说法而已。
小贴士:有时候峰型很不好的时候,不一定非得加离子对试剂,添加少许更强的有机试剂如异丙醇,二氧六环等溶剂,会起到意想不到的效果,四氢呋喃也一样,但是要说明的是,这些东西不宜加的太多,尤其是二氧六环,整个体系内0.1%就算比较多了。不过不推荐太新的新手这样。
happy王子矜
第2楼2008/12/15
看了上面,是不是觉得方法建立很简单,没什么技术含量啊,其实,只要有柱子有试剂,样品在检测器有响应,我觉得怎么也能鼓捣出来几个峰,关键是建立出来的方法,要有效,要能反应一定的情况,然后自己要对新建立方法有了解。知道它在什么情况下合适。
先说定量方法,一般来说,有面积归一法(校正/不校正)、外标法、内标法等。内标法在HPLC下适用条件不多,GC因为进样的局限性用到的多,我就不说了,如果需要可以参照外标法。
如果你打算用面积归一法定量,那你需要保证:样品里每个你感兴趣的杂质都有响应,样品和每个杂质都要分离,需要报告含量的每个杂质都要与其它峰分离。
一般如果不是客户给标准,我们要自己制订的话,那么你要保证关键的杂质的检出。不清楚关键杂质的,我告诉你一般做法。假设样品最后一步的反应是A+B→C+D,C是样品,D是副产物。那么应该制订D杂质的标准,然后可以考虑A和B中过量反应的那个,这样就给出两个杂质的标准了。一般情况下,对于纯度比较高的样品,不太会去考虑校正面积归一法。因为杂质太小,校正一下还不如修改标值方便。在某个杂质大于5%以上的时候,可以考虑校正其含量值。校正因子的算法就是把该杂质和样品定量称量放在溶解在一起进样,然后根据面积和称量的质量计算。(有客户或其它资料直接告诉更好)。
制订好标准后,我们要对方法进行验证。整套的验证方法非常复杂,也不一定都要做。具体选择什么,我按照自己的经验慢慢来说。当然,做的越多越保险——也越麻烦(不做最省事,哈哈)。其实有几点还是必须的,第一就是稳定性试验,样品配制好之后进样,然后过四个小时进样一次,不怕麻烦或有自动进样器可以每四个小时一次验证到二十四小时(我见过国外方法说明72小时稳定的),观察图谱变化,合适的方法应该保证主峰的面积相差不多,并且杂质没有增多或增大。样品在溶剂之中存放过程中可以会发生水解或其他反应,有时开发人员告诉你不太可能发生的反应,但是因为我们在配制时,样品在溶剂中的浓度非常低,也还是有发生的可能。一般情况下我们都选择了流动相做溶剂,如果有不稳定的情况发生,那么改变溶剂是第一选择。如果无法改变溶剂或改了也不行,那就要在方法里面声明,告知这个样品配制好之后在几个小时内有效。样品需要避光、低温什么的,也要写清楚。
第二要确认一下分离度,你可以选择两个不容易分离又很重要的杂质峰(或主峰),标记出来,然后规定他们的分离度在1.5以上。这一点在方法转移过程中是非常必要的。如果你的方法要给别人用,那么你在建立方法中很难分离的两个峰,有可能因为仪器系统(主要是色谱柱)方面的差异,应该分离的杂质未分离,这个时候你就必须给其他使用者提醒,避免发生不该发生的事。这个项目的验证一般是方法耐受度方面的验证,做起来很麻烦。就是把pH、温度等会影响分离度的因素改变20%,看看方法会发生什么影响。个人感觉是很脑残的验证项目,本来我们方法规定了柱温、pH什么的就是让你按这个来做的,你改了条件受到什么影响关我屁事。我们在建立方法的时候,如果试了几个体系或其他条件,对这些其实已经有印象了,五星级的推荐不做这个。就规定一下重要杂质的分离度就好。第三,其实不算正规的验证了,就是避免杂质和主峰合在一起没分离,但是你却不知道,或者样品里面还有峰没走出来,这个在建立方法的时候就要注意,有DAD检测器的可以用峰纯度看看,没有的话,最保险的方法是换个体系(最好换个性质差别比较大的色谱柱比如C18换成苯基柱)走一下看看有没有跟前面条件相比新多的杂质。没有更多色谱柱的,最简单的办法就是增大水相的比例进样,但是这样不保险。对于可能没流出的峰,可以考虑在样品主峰走完后慢慢的梯度增加有机相比例多走一会看看有没有新东西冲出。注意,比例加的太快,会有很多溶剂峰流。按照同样的梯度,走个空白做对照最好。
其他还有更严格的验证,比如验证每个杂质的线性等等,这个太复杂而且做法也很多,可以做到这一步的已经不需要看我的文章了,应该找些专业性文章或标准来看。
外标法定量需要保证:样品里要定量的峰必须和其他峰分离,要定量的峰响应要明显,并且有线性规律。一般常用的分外标一点法和标准曲线法。一点法,适用于样品含量有比较确定的范围(比如纯样品,就在95%-99%之间),我们用已知含量的标样配成浓度一致的溶液进样,然后计算。曲线法适应于样品含量范围比较大(比如回收某个东西的样品,含量可能在5%-80%之间),这时我们用标样配制几个点,比如1%,5%,10%,40%,80%,100%六个样品,分别进样,做出曲线。有样品的时候,进样,然后代入曲线公式计算。曲线建立起来后,在一个系统上可以使用较长的时间,不放心的情况下可以在测试样品前,打一个已知标样看看,结果相差不多就可以一直用下去。特别要指出的是,外标一点法在有些检测器上是不好用的,比如蒸发光散射检测器,样品的响应不是根据郎伯比尔定律,所以至少要2点定量。更多的检测器,要翻书解决,本文完全不能替代课本和专业书籍,特此声明,哈哈。
介绍完方法,然后就是如何选择的问题。这个其实是不应该我们说了算的,要看实际的需求。一般情况下,还要继续往下进行化学反应的样品,如医药或其他产品的中间体等,会对杂质很敏感,这种情况下面积归一法用的多一点。因为样品内含的杂质也许会影响到后续的反应,引起催化剂中毒或其他不可预料的风险和损失,用面积归一法可以看到各个杂质的情况。而且面积归一法定量还有它最大的一个优点,那就是稳定。在保持色谱系统一致的情况下,对于一个均匀的样品,面积归一法可以在不同的实验室得出极为相近的结果,所以尤其适合在一个企业的各个实验室之间使用。直接使用的样品,如农药或直接用来做制剂的成品药,用外标法的比较多,因为大家做出来后要使用的,所以需要的实际的质量含量(w/w)。外标法反应的就是样品内的实际含量,而不像归一法只是根据面积得出来的纯度。其实现在更多的情况是两种方法都报告,所以最好能用一张色谱图出两个结果,把两种方法要注意的都照顾到。
我这里说的只是新建立方法简单的自己要做的定量和初步验证,真正的方法验证是一个很严格的程序,建议参考我们的药典或是FDA等其他相关资料。上面举的例子只是为了说明问题,比如曲线点浓度的选择等等,不要简单的照搬,还是要按照需求来。验证方面的东西很多很复杂,各个机构公司要求也略有不同,希望大家把在工作过程中遇到的问题或感悟拿到我们仪器信息网来,我们一起讨论分享,共同提高。