

东风恶
第2楼2010/03/30
而对于制剂而言,由于原料药的质量标准已对各杂质进行了控制,在制剂的质量标准中仅控制降解产物,故在《新制剂的杂质研究指导原则》中规定:在制剂质量标准中任一单个非特定降解产物的限度不得过鉴定限度(见表2)。
表2:制剂的杂质限度
根据以上规定可以引申出:在仿制药或改剂型药品以及药品上市后变更等的研究中,即使出现了新的杂质,只要新杂质的含量低于表1或表2中的鉴定限度,就可以认可这些新杂质的安全性。
从以上指导原则的规定,也可以看出:现在国际上对非特定杂质的限度要求是比较严格的。而我国在这方面尚处于起步阶段,仅一些质量控制比较严格的质量标准才收载有对非特定杂质的限度要求,且具体限度远远大于鉴定限度,也不考虑临床用量,只一般性地规定为“任一单个杂质不得过0.5%甚至1.0%”。从保证药品的安全性与从严控制产品质量的角度出发,我国应加强对特定杂质的控制。希望本文能引起有关各方对非特定杂质的关注,使我国在这方面的要求逐渐与国际接轨,以利于我国的制剂成品走向欧美等国际市场。

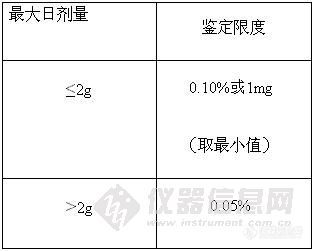 表1:原料药的杂质限度
表1:原料药的杂质限度































