

guizi1985
第2楼2011/06/18
我试过几个方案
1.因为其中有一种是酸性药物,所以用酸化乙酸乙酯(三氯甲烷)8ml提取,8000r/min离心10min,残渣重复提取一次,合并上清,正己烷去脂,40度氮气吹干,1ml流动相洗残渣,PSA 0.1g去色素,上机检测。(用三氯甲烷也试过,乙酸乙酯可以将药物提取出来,但是杂质也全部提取出来了,三氯甲烷相比乙酸乙酯提取干净很多,但提取效果没乙酸乙酯好)。
2.由于本实验做肝脏采用1%偏磷酸40%甲醇提取2次,提取回收率均可达80--90%,本实验室土壤中用1%偏磷酸40%甲醇20%乙腈提取均可达80--90%。所以
3.由于粪便中1%偏磷酸提取不出来那个酸性药物,所以试过2%、3%、4%的偏磷酸,2%就可以提出来,所以采用了2%偏磷酸40%甲醇20%乙腈10ml提取,正己烷去脂,硫酸铜沉淀蛋白,PSA去色素,
4.用2%40%20%提取后图谱分离度不好,基线不平,难以计算回收率,所以采用液液萃取的方式净化,用乙酸乙酯反萃取2次,吹氮气,流动相溶解,上机检测。
可是图谱:
基线仍然高,杂志干扰大,分离度差, 个别药物回收率 还是很低。
图: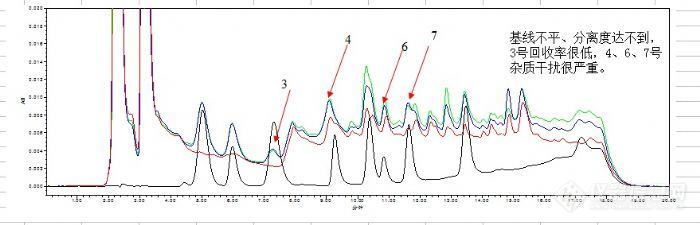
guizi1985
第4楼2011/06/18
这根柱子柱效不怎么好了,因为样品脏,不敢用好柱子,所以就先用废柱子跑了。至于流动相 我们当时摸索了很长时间,关于药物的梯度洗脱条件试过很多种方法, 好不容易将8种药物可以完全分离出来了(标样)。
其实我更想问的是1.怎样将8种药物 全提取出来?(因为试过很多试剂 回收率一直不怎样
2.基线怎样降低到跟标准品的基线差不多)
基质内源性干扰很大,是否可以用大孔树脂 吸附杂质?
图中前3种药物 出峰时干扰不是很大,后面5种时 干扰就多了,是不是基质中非极性杂质更多些?怎样针对性去除?请大家帮帮忙分析下好么?
guizi1985
第9楼2011/06/19
嘿嘿~谢谢鼓励!
1.这8种药中第三种药物标准品就是相当难容,在弱碱性条件下溶解度还好些,还有就是第6个药,出峰时总有较大的干扰峰,在我试过的有机溶剂中都会出现那个比药物峰还高的杂质峰。
2.我的进样体积:40ul,估计进样量确实大了,
3.呵呵。。。柱子就是普通的C18柱,
我有疑问:您说可适当加三乙胺封闭未键合的硅醇基,改善峰形,提高分离度。三乙胺浓度大约0.1%。这个具体怎么操作呢?我的流动相是乙腈:0.5%甲酸水
guizi1985
第10楼2011/06/19
我还试过另一种方法,跑的图如下: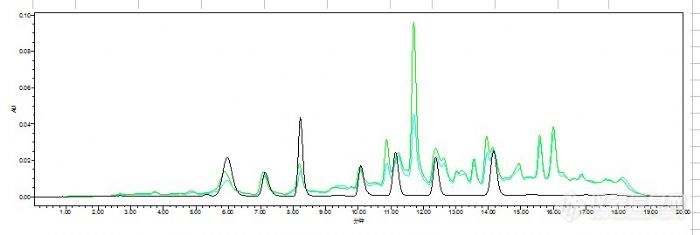
这是用2%偏磷酸20%乙腈提取两次,用三氯甲烷萃取提取液后的结果,三氯甲烷萃取会干净很多,但是后面4种药物杂质仍然分不开,主要是后面4种。第1和2回收率还是达不到要求。

































