

你一见我就笑2169
第4楼2018/07/18
非常感谢你的回复。
1、我做的确实是研发公司的新产品方法开发,有时候客户提供方法,会遇到和自己的方法有差异的情况。
2、很多产品都处于打通路线和优化工艺阶段,存在很多变数,因此我们分析在研发确定工艺路线之前也没有及时做相关的方法验证工作,所以经常会在后续的检测过程中发现一些问题,再临时做调整的情况。
3、DAD看纯度的工作我们都看的,但本人发现若包在主峰里的杂质较小时,纯度也是通过的。但也没有相关数据表明,包含的杂质超过多少可以通过DAD纯度的结果体现出来……尽管DAD纯度检测不是百分百准确,但至少能说明一些问题。顺便请教一下:如果DAD纯度检测发现未通过,该如何优化呢?(梯度拉长,看看是否有新杂质峰与主峰分开,再次检测主峰的纯度?)
4、关于重复性和溶液稳定性我们也做。顺便请教一下:如果某样品溶液重复进样发现纯度有差异,如何界定到底是方法重现性的问题,还是溶液稳定性的问题?
5、选择不同浓度的样品检测,是为了查看线性?还是通过此办法判定样品是否以两种形态存在?(我可不可以理解为:样品浓度较高时,有可能在体系里没有完全转化成一种形态,浓度较低时,基本上可以完全转化,以此来作为判定依据?)
6、请教一下,如何查询某一化合物的PKa值,以便为选择合适的流动相PH值作为依据?我们多数是查不到这个数据的,只能通过结构简单判断一下其酸碱性,再选择一个合适体系(比如酸性体系)进样。经常会遇到理论和实际不一致的情况,那就需要再尝试另一个体系(比如碱性体系)了,基本上能满足绝大多数产品的测试。但是个别产品不保留或强保留或峰形不好,就只能再更换缓冲盐的酸性体系或中性体系或直接选用亲水性色谱柱测试了,这样工作量就比较大了。请教一下,有没有相关的经验分享?
你一见我就笑2169
第6楼2018/07/18
归一化法。
1、我个人理解的是:如果排除了样品溶液稳定性问题、重复性的问题、线性问题、不同形态的问题,理论上来讲不同的方法,结果应该是差异不大的,结果差异大就考虑杂质少的那个方法可能某些杂质重叠的原因,这样理解有没有什么问题?
2、在方法开发初期,尚未做方法验证阶段,如何根据化合物本身的结构和性质,尽可能准确的选择一个方向(酸性体系?碱性体系?缓冲盐体系?普通的C18柱子?亲水性柱子?专用柱子?)?有没有相关经验分享(尽可能减少前期方法选择不合适,后期再重新换方法的麻烦)?
浪淘沙隐
第8楼2018/07/18
你一见我就笑2169
第9楼2018/07/18
非常专业,再次感谢你的耐心解答。
1、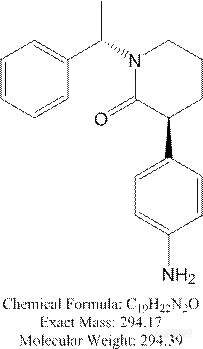 做该化合物的分析过程中,遇到了你说的类似问题:
做该化合物的分析过程中,遇到了你说的类似问题:
酸性条件下(A:0.1%磷酸水溶液;B:乙腈 运行梯度),未见明显杂质,纯度较高,光谱纯度也是通过的,见下图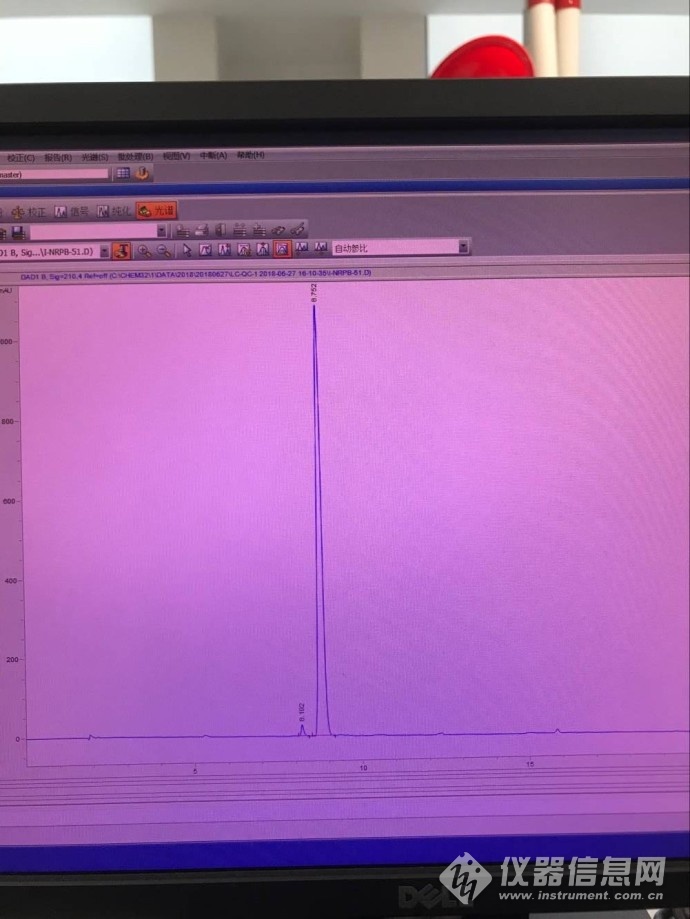
换做碱性条件(A:0.1%氨水溶液;B:乙腈 运行梯度),纯度较差,主峰后面出现了18%的峰,且光谱不一致,见下图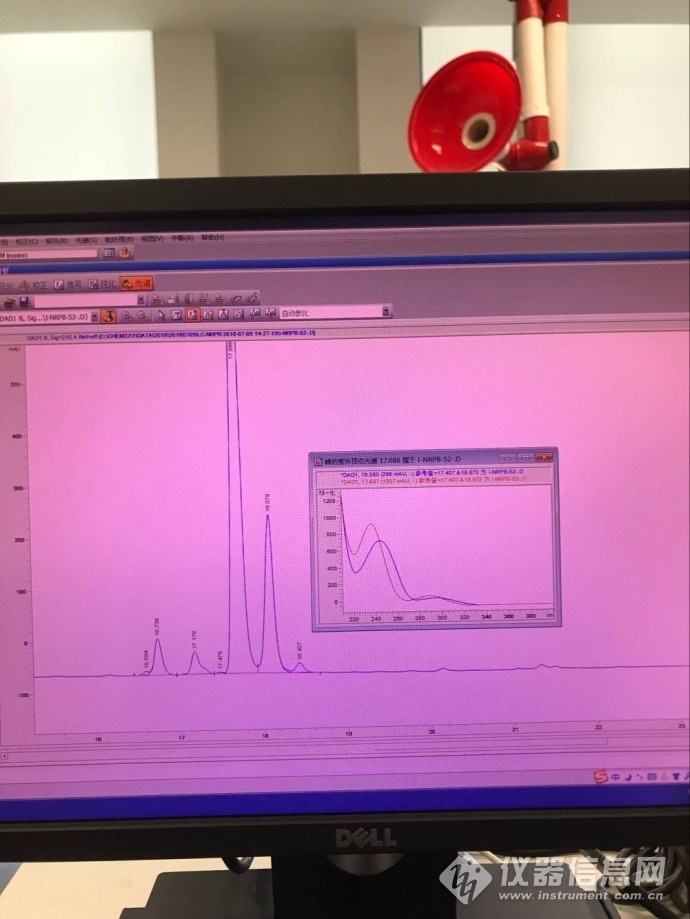
通过改变条件,效果均不佳,始终与主峰达不到完全分离;重复性还不错,微小差异,怀疑是分度不完全引起。
我的怀疑和你遇到的情况一致,怀疑是某一种两种形态的存在,但没有十足的把握,始终不敢轻易下结论,打算后续继续想办法进一步证实这个怀疑。
2、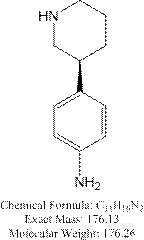 该化合物,在酸性体系中,基本无保留;碱性体系中,峰形较差(展宽、对称性差),见下图:
该化合物,在酸性体系中,基本无保留;碱性体系中,峰形较差(展宽、对称性差),见下图: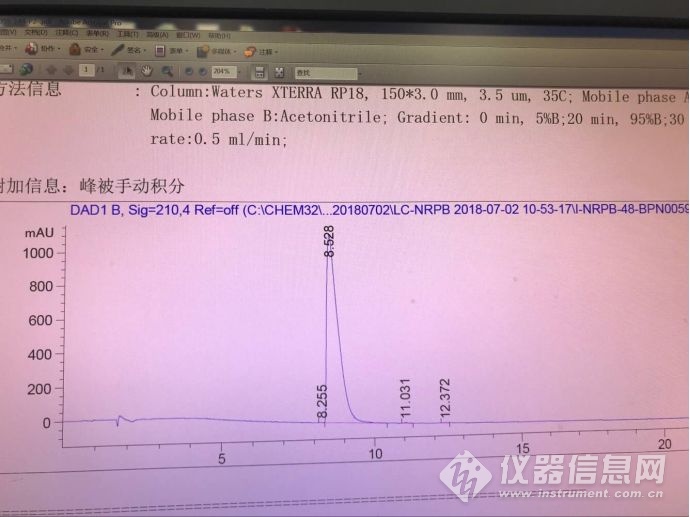
行业通病——钱少或多,还没抽出时间继续换条件尝试……
3、上述两个问题,都是我最近的一个项遇到的,身为同行,希望与你分享。只是目前还没有完美的解决方案,后续有了结论,我再进一步回复。

































