

(一)适合HPLC方法开发的化合物
(二)反相色谱出峰原理
样品分子在极性流动相和非极性固定相之间进行分配,疏水性强(极性弱)的化合物保留强,后出峰;亲水化合物(强极性)保留弱,先出峰。
引用:使用C18色谱柱的反相方法,出现未知峰时,可大致判断该未知物质极性的强弱,给技术员参考性的建议。
官能团极性大小:
饱和烃<烯烃<芳烃<有机卤代物<硫化物<醚<硝基化合物<二甲胺<酯类<醛<酮<硫醇<胺类<亚枫<酰胺<醇类<酚类<羧酸
(三)反相色谱条件选择
(1)各个色谱参数对分离的作用
色谱参数 | 对分离的作用 |
波长 | — |
溶液配制-稀释剂的选择 | 需要与流动相互溶,并且需注意溶剂效应 |
流速 | 影响不大 |
柱温 | 影响小,但对某些特殊结构有较大影响 |
流动相 | 起主要作用 |
固定相(色谱柱) | |
洗脱程序 |
(2)反相色谱中波长的选择
第一,对目标化合物进行全波段扫描。
第二,选取的波长需大于流动相的截止波长,反相中若用了缓冲盐,则最低205nm。
第三,含量的方法中,波长尽量选取较为平滑的峰顶或峰谷。
第四,有关物质方法中,波长需对比各个杂质及主物质在同浓度水平下的UV曲线,选取各个物质E值相当的波长,并需确认校正因子。校正因子在0.9-1.1之间以1.0来计;在0.2-5之间,校正因子法能得到较为准确的结果;若校正因子大于5或小于0.2,则需用外标法来进行计算。
(3)反相色谱中稀释剂的选择
①溶剂强度顺序
水<甲醇<乙腈<乙醇<四氢呋喃<丙酮<二氯甲烷
其中二氯甲烷和水不互溶,其余和水互溶。
乙腈/甲醇/四氢呋喃和水不同比例的溶剂强度如下图
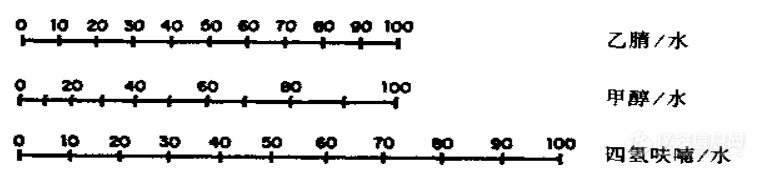
②溶剂效应
a.如何判断是否是溶剂效应呢?
稀释剂的溶剂强度大于流动相的溶剂强度,可能导致峰分裂。降低进样量,峰形明显趋向成为一个或者成为了一个峰,此为主要判断依据;或者用DAD扫描,分裂的两个峰紫外吸收曲线明显一致,此为辅助判断,因为很多分离不好的化合物的结构是接近的,紫外吸收曲线可能相差不多。
b.如何避免溶剂效应呢?
选取稀释剂时,溶剂强度应小于或与流动相初始比例的强度相当。或者在允许范围内,降低进样量。
稀释剂不仅需要溶解样品,更需要与流动相互溶。
c.溶剂效应裂峰实例


③稀释剂选择考虑因素
例1:稀释剂pH
如果用正己烷溶解了样品,在反相色谱中运行时,会与水形成液珠,可能会伤害仪器及色谱中。
有时候也需要注意稀释剂的pH,若样品中某些物质对pH很敏感,需要将稀释剂pH与流动相的pH保持一致。
例2:稀释剂稳定性-稀释剂不合适
利伐沙班某一步中间体中,流动相A为0.1%磷酸水,用乙腈-水去溶解样品时,其中一杂质会慢慢降解,最终选用乙腈-0.1%磷酸水作为稀释剂,溶液有较好的稳定性。
PS:对某些有卤素基团的化合物,需注意其是否在稀释剂中能稳定存在。
例3-1:稀释剂稳定性-温度
布瓦西坦某一步中间体中,脂肪上连接了I,经过大量实验发现,即使在纯乙腈中,室温下I也极容易掉落。最终采取稀释的方法是,将乙腈在0℃左右保存,来样后,马上用0℃的乙腈稀释,并且样品盘控温5℃。这样才抑制了样品的降解。
例3-2:稀释剂稳定性-温度
阿托伐他汀钙缺陷,需要路线前沿,某一步原料的杂质结构式苯环上连接了两个Br,在低浓度下,Br容易掉落,导致LOD,LOQ一直做不过去,最后将样品控温5℃后,才将方法确认做完。
总结:溶剂效应,与样品及流动相的溶解性,样品溶液的稳定性。
(3)流速
在色谱柱耐受的压力范围内就行,对分离影响很小。
(4)柱温
一般情况下,柱温的升高或降低,可能会改善分离,也有可能会降低分离。不要将柱温升高至临近柱子的耐受温度,尤其是在缓冲液pH6.5以上时;pH2-3时,有的色谱柱如Agilent SB系列可以耐受80℃的高温。
对于特殊的结构,柱温的变化会有显著的影响
如酮式烯醇式互变的结构。在低pH,高温下,有利于趋向酮式结构,在高pH,室温或者低温下,有利于趋向烯醇式结构。所以开发方法时,酮式烯醇式的结构需要去了解以哪个结构为主要存在的,改变柱温及缓冲液pH让其绝大部分以同一形式存在。
柱温需考虑:柱子耐受,某些特殊结构需要合适的柱温。
(5)流动相的选择
①流动相中的有机相选择
首选乙腈,乙腈-水流动相体系,可用于低波段(185nm-210nm)UV检测,乙腈水的流动相粘度非常低;
第二选择是甲醇,对比乙腈的优势是价格便宜,对缓冲盐的溶解性稍好于乙腈,但是甲醇截止波长205nm大于乙腈,而且甲醇水体系粘度大,在甲醇-水=1:1时粘度达到最大,柱压较高,需注意色谱柱是否能耐受;
最后考虑THF,THF在UV中有吸收,易氧化,更换色谱柱后平衡较慢,但是对比乙腈甲醇,有独特的选择性,对于某些不好分离的化合物,可以尝试加入少量THF。
混合有机相具有独特的选择性。
②流动相中的缓冲液选择
了解目标物的酸碱性及pKa,将缓冲液pH调节至大于或小于1.5pKa;
若是酸性化合物,pH小于其pKa会增大保留;
若是碱性化合物,pH大于其pKa会增大保留;中性化合物,pH对保留影响较小。
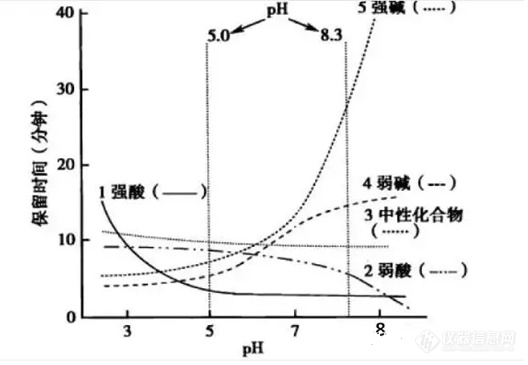
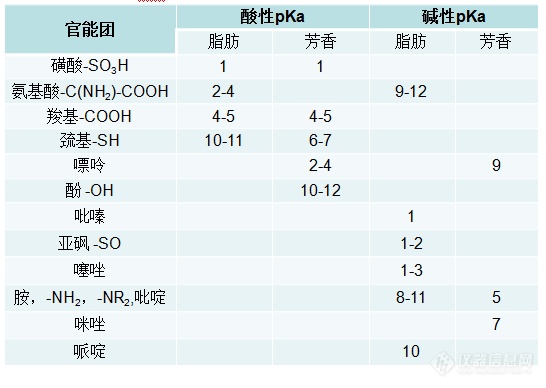 b.HPLC中常用的缓冲试剂
b.HPLC中常用的缓冲试剂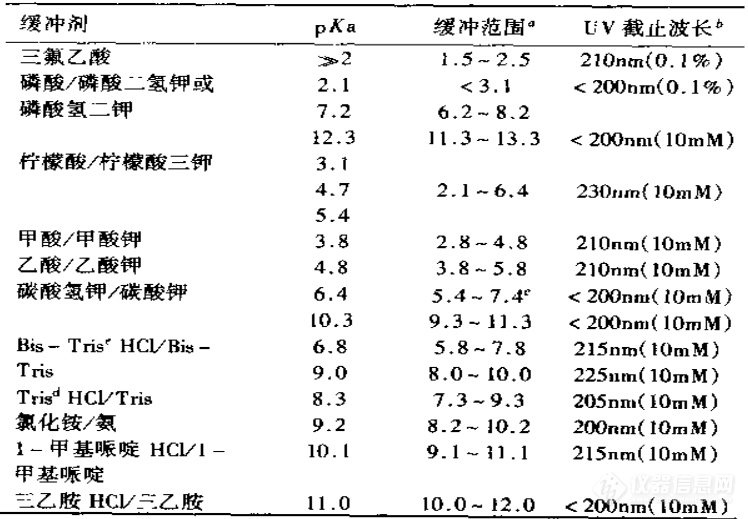
c.案例
例1:流动相缓冲能力优化案例
氨基酸盐酸盐化合物含量方法中, 缓冲液用的是5mM磷酸二氢钾, 样品中氨基酸的峰和标准品中氨基酸的 峰未能完全重叠在一起。如图所示:
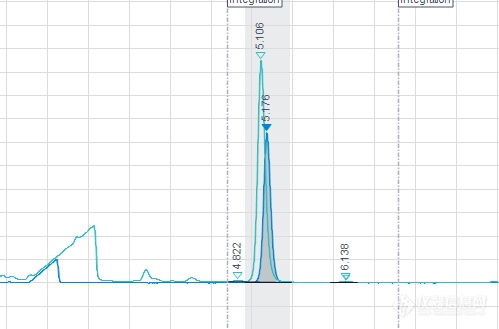
后期将缓冲液换成5mM磷酸二氢钾和 5mM磷酸氢二钾的混合液,能完全重叠,如图所示:
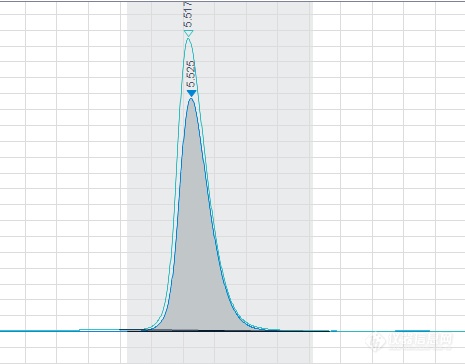
原因分析:5mM磷酸盐pH4.6左右,无 缓冲能力,而两者的混合液pH6.95, 缓冲能力强。
例 2:缓冲盐析出问题案例
曾近我开发过pH7以上10mM磷酸盐缓冲液的方法,在与有机相比例达到50:50时,柱压会缓慢上升,所以遇到此类转移过来的方法,需要时刻注意柱压,若是发现每一针都比前一针压力都高,则方法需要调整,不然柱子容易损坏。
最后,将方法pH降低至6.5,用三乙胺-磷酸水的体系解决了该问题。
d.缓冲液需考虑的因素:
缓冲液pH,需大于或小于化合物1.5pKa。
UV的吸收,需大于缓冲盐的截止波长。
缓冲液和有机相的溶解度,在方法中最高有机相+5%比例下,缓冲盐未析出。
缓冲液浓度,一般5-25mM之间。
缓冲液的稳定性,比如我们很少用到碳酸盐去作为缓冲试剂,因为碳酸盐缓冲液放置后,CO2会逸出,会导致pH升高。
缓冲盐缓冲范围,若影响较大,则需尽量将pH调整在缓冲范围以内。
对HPLC系统的腐蚀,比如盐酸会对整个系统造成损伤。
(4)反相色谱中色谱柱的选择
①常用色谱柱类型
C18色谱柱 | 比较常用 |
C8色谱柱 | 相比于同类型的C18色谱柱,相同条件下,化合物保留降低 |
苯基柱 | 适合区分共轭双键的化合物 |
HILIC色谱柱 | 反反相系统。适合亲水,在反相上保留很弱的化合物 |
离子交换色谱柱 | 适合保留弱的酸碱性化合物。如含有SO32-、COO-、NH3+等离子化合物 |
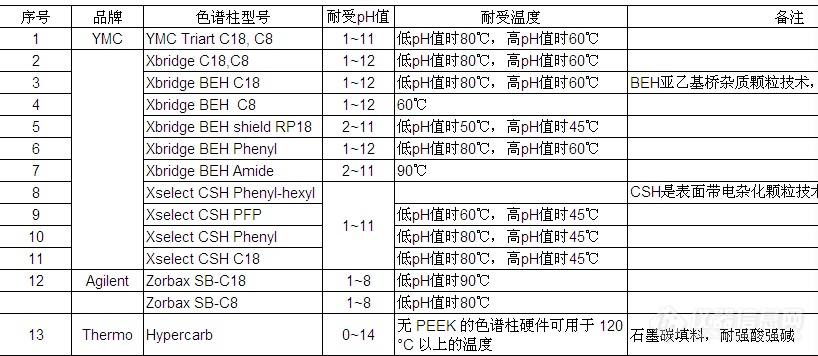
②不同耐受功能色谱柱品牌推荐
a.耐水性

b.耐高pH

c.耐高温
③固定相孔径的选择
根据分子量选择合适的孔径
 ④色谱柱长度及粒径的选择
④色谱柱长度及粒径的选择N正比于长度,反比与粒径的二次方。理论上说,粒径越低的色谱柱柱效越好,但是压力也会越大,所以综合考虑,HPLC上色谱柱粒径基本选用3μm-5μm之间。
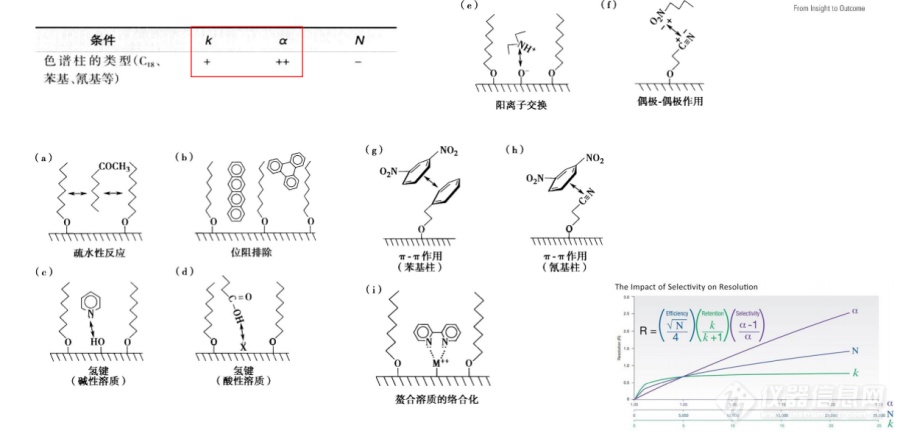
⑤固定相的选择性
(四)方法开发的步骤
首先使用封端,高纯硅胶,C18色谱柱,如Agilent XDB C18,Waters Xbridge C18,YMC Triart C18色谱柱等,开全波段扫描,选取合适的波长。
了解目标物的pKa,选择合适缓冲液,将缓冲液pH调节至大于或小于1.5pKa。
第一种方法-等度调节:将有机相调至80-100%,等度洗脱,然后慢慢调低有机相比例,直至目标物之间达到分离。
第二种方法-梯度调节:初始梯度调节为50%有机相,以5%/min比例升至80%或者100%,观察目标物之间的分离,调节初始梯度及有机相百分比的速度来达到各物质分离。
若不能将目标物完全分离,可观察哪几个结构的化合物无法分离。
情形 | 解决方法 |
中性化合物和含有极性官能团的化合物未分开 | 调节缓冲液pH |
位置异构体未分开 | 更换杂化颗粒色谱柱,或者PFP色谱柱 |
共轭双键化合物未分开 | Phenyl色谱柱,PFP色谱柱 |
若需要减弱保留时间 | 使用C8色谱柱,使用粒径更小的色谱柱。 |
酸性化合物保留弱 | 一、更换为耐受纯水的色谱柱,使用100%低pH缓冲液去增大保留 二、使用阴离子交换色谱柱 三、缓冲液中加入阳离子对试剂,如四丁基硫酸氢铵 四、HILIC模式:对于亲水性的化合物有较大保留。 五、观察是否有可以衍生的官能团。 |
碱性化合物保留弱 | 一、使用阳离子交换色谱柱 二、缓冲液中加入阴离子对试剂,X烷磺酸钠 三、HILIC模式:对于亲水性的化合物有较大保留。 四、观察是否有可以衍生的官能团。 |
酮式烯醇式互变,以酮式为主要存在形式 | 使用低pH下耐高温的色谱柱。将柱温调高,观察峰是否成为一个。 |
有机相也可以改变化合物的分离
选用合适的缓冲液和固定相,改变有机相,乙腈,甲醇,不同比例的乙腈甲醇溶液,或者往有机相中加入1-10%THF。
实例:
阿托伐他汀钙缺陷中,前沿起始物料的方法开发,用磷酸-三乙胺水溶液作为缓冲液,甲醇-乙腈(60::40,v/v)作为有机相,成功的分离了8个化合物,并且通过了carry-over验证。
研发分析技术交流与学习,请关注微信公众号“研发分析之路”

































