

火焰原子吸收法测定金矿样品经验分享
接触火焰原子吸收法测金已经有7个年头了,期间也遇到过很多问题,从中也不断学习和积累了一些浅见,供大家参考。原子吸收法测试的影响因素很多,有气体方面的,有仪器状态方面的,也有标准溶液方面的,还有样品前处理方面的;在论坛也看到很多新手就以上这些方面发出过求助帖子,下面就这几个方面一一阐述,不足之处请大家批评指正。
一、气体方面
气体主要有燃烧气和辅助气,燃烧气大多用的是乙炔和笑气,我用过较多的是乙炔;辅助气用的是空气。燃烧气和辅助气之间的各种情形,安老师已经给大家分享了,我就不班门弄斧了。我这里要强调的是,乙炔纯度一定要高纯的,

否则,你看到的火焰颜色不是蓝色的而是桔红色的,

这还不要紧,要命的是,你好不容易画出了工作曲线,没过多久(也许就分析完20个样品),你的QC(质控标准)就偏高很多了,比如说你的QC正常值是5.0,偏差允许范围是2%的话,那你的值范围就是4.9~5.1之间,但是实际上你的QC测得值已经是5.5或者更高了,然后你观测火焰,发现火焰很不稳定,像是有风在吹着它似的;如果你熄火拆下燃烧头,很可能上面已经有黑黑的积炭了。我清楚记得刚接触的时候,以为点着火能测试就行了,其实不然,碰到过连续换了3瓶气都是这样的,怎一个郁闷了得。所以说,乙炔必须得是高纯的,千万别贪便宜,不然,受罪的就是你。再说辅助气,空气压力要达到分析要求,不然可能点不着火的,空气通常是用空气压缩机来获得的,这里也要提醒大家,湿度大的时候要给空气压缩机排排水,空气压缩机不能用有油的哦。
二、仪器状态方面
仪器状态好不好,测试人员最有发言权也是最了解的,状态好的话,测试起来得心应手,一大批样品很快搞定,结果是准确可靠,那叫一个高效率;状态不好的时候,测试的结果自己心里都没底,几个样品够你折腾大半天的。那可能有人要问了,什么样的状态才叫好?
引用国标中的一个判断条件:

简单的来说:就是工作曲线的线性要好,不说0.9999,至少也是0.99以上吧,其次是QC结果在合理控制范围内,如果标准物质的结果也跟参考值吻合的话那是再好不过了。请看下图
工作曲线:
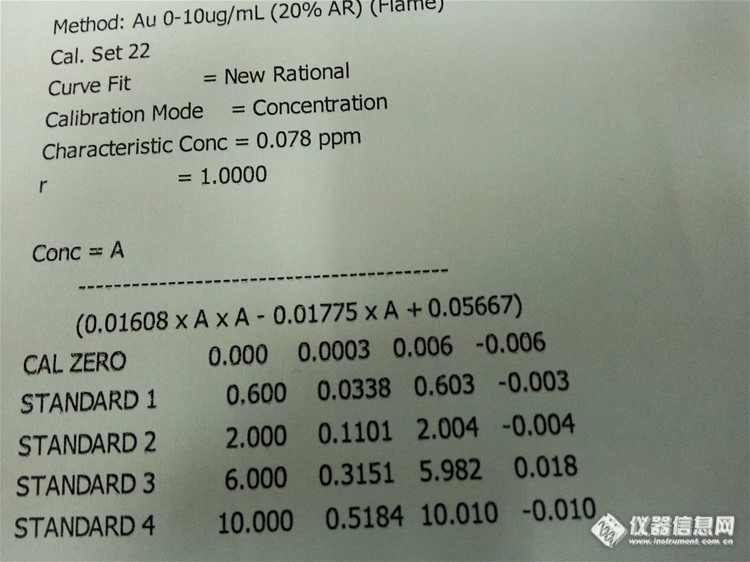
测试数据(删除一些数据,重点看QC)。
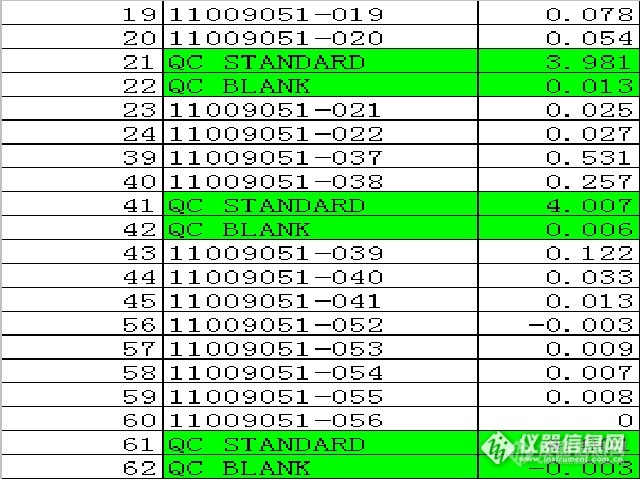
那状态不好是什么情况呢?很可能你的工作曲线线性很差,画完曲线测QC样品都显示超出范围或者Wrong(错误)。这时候你需要进行仪器的调节,优化状态或者清洗、清洁仪器光路、燃烧头、雾化器等。
看下图数据蓝色部分为重点关注区域:
工作曲线:

测试数据(看波动、振幅大小)
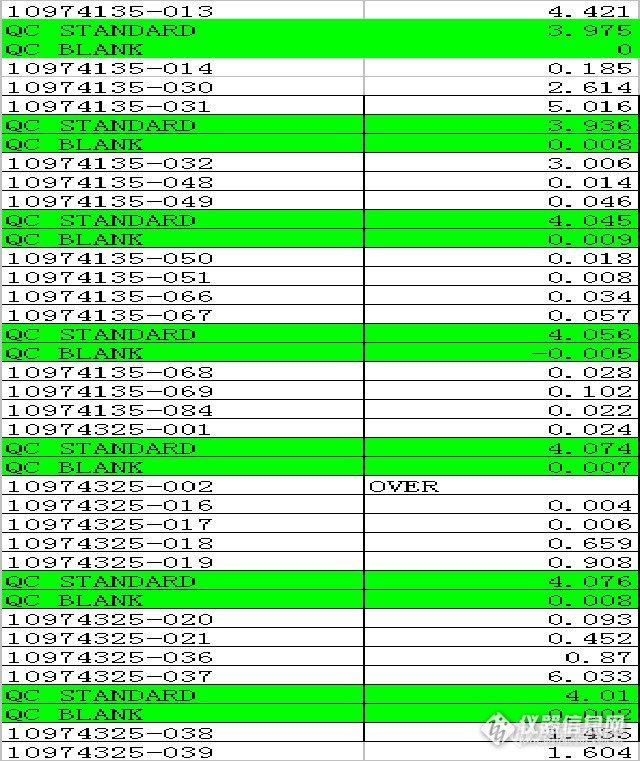
还有就是,点火前要确认乙炔和空气的压力是否达到要求的压力,否则要么点不着火(空气压力不够);要么火焰很不稳定,并逐渐变瘦(乙炔压力不够)。
三、标准溶液方面
说起这个有人会不以为然,有时候你曲线做的很漂亮,QC也很好,但是测试标准物质结果就是偏高或者偏低5%,在排除了前处理、仪器状态、气体方面的情况下,你是不是该怀疑一下你配制的标液是否有问题?你进行过比对或者验证吗?是和上次的还是外购的标准溶液比对?我们有一次,我休假几天,刚好溶液用完了,一个同事帮忙配制的标准溶液系列,测试的时候没来及验证,结果测试结果比往常高了3%(通过标准物质看出来的),最后证明是标准溶液整体配低了才导致结果就相对偏高了。所以说,配制标准溶液也是检验一个分析检测人员的个人技能的一个手段。
四、样品前处理
如果是严格按照标准方法或者企业内部经过验证的方法来进行处理的话,一般是不会有什么问题的;但是有人员做事不高仔细认真,或者叫不够严谨的话,或许体积就多了(或者少了)那么一点点,这样溶液的浓度也就不一样了,看似很细微的差异,放大就很厉害了。拿我们的金溶液来说吧,通常最后体积是控制在4ml,但是如果你多了0.2ml,那就是5%的偏差了,那你还拿什么来保证你结果偏差在5%以内呢?检测人员在拿到样品溶液的时候最好先确认一下体积是否正常,如果相差太大就有问题了,这样测出来的结果你还觉得可信吗?
小结
通过以上几个方面的絮叨,或许对你遇到的类似的情况可以提供一些参考和借鉴,快速找到解决问题的方法和途经。以上是个人的经验之谈,不足之处,还请专家和老师补充和完善。

































