气相色谱在日常应用中大家普遍将注意力集中在色谱柱、检测器等等关键部件上,常见的样品分析条件设置也多与它们有关。但是同样重要的进样口维护却常常被大家忽略,尤其是刚刚接触气相色谱的初学者,很多问题的产生往往和进样口的条件设置有关,而进样口中小小的衬管则是许多问题的核心,今天我就来说说这小小衬管的大学问。
气相色谱称管的日常保养与维护:
常见与衬管有关的问题有:
① 衬管的内壁被污染,或有进样隔垫碎渣,易导致分析结果重现性差(如保留时间、峰面积等的重现性)或出现鬼峰;
② 衬管内的石英玻璃棉等装填物填装不当,或装填物、衬管内壁有活性点,易导致实验结果重复现差。
③ 气相色谱仪长时间运行后,进样口经常残留有凝固的样品。
当进样口有样品残留时,a.如果凝固样品残留在进样隔垫上面或下面,易导致进样器进样时针尖不好插入;
b.如凝固样品残留在分流管路部分内部,会导致管路内径变细,甚至堵塞;
c.当进样口温度比通常工作温度高很多时,之前凝固样品可能会出鬼峰,影响分析结果的重现性。
因此,衬管和进样口要定期保养。
气相色谱衬管的日常保养方法:
虽然分流/不分流进样口,填充柱进样口和大口径毛细管柱进样口,对衬管的结构要求不一样,但是不同结构的衬管的保养方法基本一样。
为降低因衬管和进样口被污染给测定结果带来的影响,应经常对衬管和进样口内部、进样口过渡接头等进行检查、清洗,必要时要更换衬管。
对于衬管和进样口的保养,一般分为日常保养和定期保养。下面分别介绍:
衬管的保养:
① 衬管的日常保养
清洗衬管,是常用的保养方法,适用于衬管内壁被污染或有进样隔垫碎渣。
首先,从仪器中小心取出衬管,用镊子或其他小工具小心移去衬管内的残留杂屑和装填物如玻璃毛,移取过程不要划伤衬管表面。
先将衬管上端的石墨压环(或硅橡胶环)卸下,这上面易附着溶剂,会导致鬼峰。将衬管泡在有机溶剂(如丙酮)里,然后,用蘸有溶剂(如丙酮等)的纱布擦洗衬管内壁。
如果衬管内壁上的严重污垢部分擦不掉,可将衬管浸于有机溶剂(如丙酮等)中放置数小时,直到能用蘸溶剂的纱布擦净内壁。
最后,烘干(或吹干)衬管,装入新的填充物,装好衬管。
② 衬管的定期保养
衬管内的装填物填装不当,或装填物、衬管内壁有活性点,可能导致分析样品被吸附或分解,直接引起实验结果重现性差。如衬管或装填的石英棉活性过高时,分析有些农药样品的色谱峰会重现性差,结果也会有很大的偏差。
一般分析极性样品时,要及时更换或硅烷化衬管和装填的玻璃棉。衬管和玻璃棉的DMCS(二甲基氯硅烷化)处理,可如下操作:
首先,将衬管、石英棉用丙酮等有机溶剂清洗,晾干后在5%DMCS/正己烷中浸泡一夜。然后,取出浸泡后的的衬管、石英棉,立即用甲醇清洗2-3次,然后在甲醇中再浸一小时左右。浸泡后取出,晾干后,装好衬管。
实际案例
测定农药甲胺磷。
gc450气相色谱仪,PFPD检测器;进样口250℃;检测器300℃;柱温程序升温,80℃保持1分钟,以20℃/min升至180℃,再以5℃/min升至230℃,再以15℃/min升至250℃,保持7min。进样后谱图异常(见图1),约6.4min处的主成分峰甲胺磷峰太小。关机后,对仪器进样口清洗后,没有解决问题;再取出衬管,发现衬管和玻璃棉都很脏,清洗,并超声玻璃棉后,装好衬管,开机重新进样,得谱图2,主成分峰甲胺磷正常,谱图恢复正常。

什么时候应该更换称管了?
衬管一般由玻璃或石英材料制成,它的型号很多,适用于不同类型的进样口。衬管是进样口的核心,样品在此汽化,随着分析样品次数的增多,衬管会变脏,此时若通过清洁衬管等方法不能解决衬管导致的问题,就应该考虑更换衬管。
影响衬管寿命的因素通常是:
① 样品的性质;
② 进样口的温度;
③ 仪器的日常保养;
④ 使用不当导致破损。
长时间使用后,未挥发的组分滞留在衬管内,衬管会变脏;当衬管内的污染物积累到一定程度时,会直接影响分析结果。如导致分析结果重现性差、色谱图峰形前伸、拖尾、峰分裂、出现鬼峰等现象。衬管破损会导致分析结果重现性差,甚至不能正常分析。
对于衬管,根据日常工作经验,如果近期分析的样品组成复杂、高沸点组分多,因考虑到样品的性质,进样口温度不是设置很高,实验次数频繁而没有及时的维护衬管,衬管可能会积累污染物;如果不维护而直接去做实验,可能导致仪器工作时的色谱特征不正常,如出现分析结果重线性差、色谱图峰形前伸、拖尾、峰分裂等现象。如果多次维护衬管后,仍不能解决上述问题,可更换衬管。
实际案例
在一次气相色谱分析中,应用Agilent6890N的序列,自动进样走了几针后,仪器出现故障自行停止;面板“status”显示柱前压为0.4psi,而设定压力为7.1psi,载气为氦气,柱内流量1mL/min,分流比40︰1,初步判断漏气;增大柱内流量为2mL/min,发现进样口压力提高,为0.6psi,判断有漏气。
关仪器后,对色谱柱的两个接口各自拧1/4圈,压力没有提高,可判断色谱柱前后接口处无严重漏气;打开隔垫上的螺帽,取出隔垫,用食指堵住进样口,发现柱前压力恢复正常,故判断为进样隔垫漏气。其原因可能是实验室气相色谱的自动进样器和顶空进样器常交替使用,而顶空进样针有时会向四周转动,引起隔垫提前损坏。更换隔垫后,柱前压恢复正常,仪器正常工作。
进样体积是不是超过衬管反应室容积了?
进样体积不当是造成精密度差的原因之一。分流进样方式的进样口流速比较高,样品流出进样口比不分流进样方式快得多。因此,分流进样时的进样体积,一般不会超过进样衬管反应室的容积,因此分流模式对进样体积要求没有那么严格。然而,不分流进样对进样体积要求要严格的多。怎么判断分流/不分流进样口的进样体积是否合适呢?
衬管的容积,是影响定性定量分析结果的重要参数之一,通常要求衬管容积至少等于样品中溶剂汽化后的体积。如果衬管容积太小,而进样量很大,可能引起汽化样品“倒灌”进汽化室,进样时柱前压会突然升高;如果衬管容积太大,可能使样品初始谱带展宽。
在常规色谱条件,一般,进样体积大于1μL时的进样的重现性不好。因为衬管的容积有限,当进样的体积很大而进样口温度很高时,样品的膨胀体积会超过衬管的有效体积,样品蒸汽“倒灌”,从隔垫吹扫气出口出去,造成进样的重现性变差。
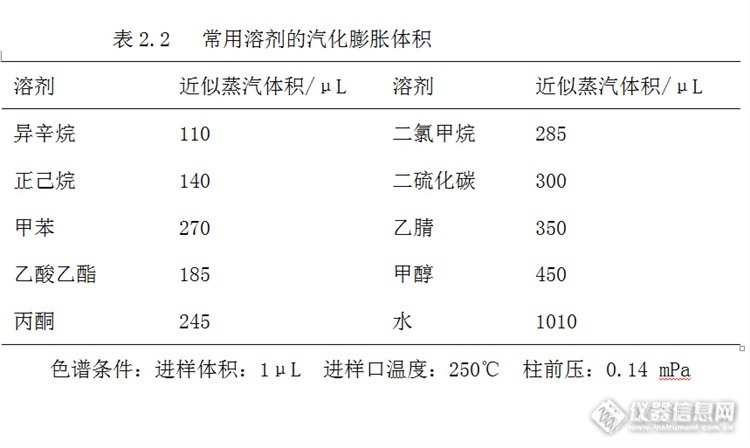
这个问题的解决取决于样品的溶剂类型和衬管的容积。建议使用蒸汽体积计算器,针对化合物的性质和汽化室的特点,估算最终的扩散体积。
常用溶剂气化膨胀后的体积,见表2.2,在选配衬管的结构和容积时可以参考。

实际案例
色谱条件:用酸改性聚乙二醇(20M)毛细管柱,柱温140℃,汽化室温度190℃.载气(氮气)流速为每分钟60mL;自动进样器,重复5次进样。该试验条件下检测磷酸三丁酯的含量。
其他条件不变,只改变进样量。进样量为1.0μL时,样品峰面积较小,峰面积的相对标准偏差较大(4.9%),谱图见1。为提高分析的精密度,尝试加大进样量以减少测定误差。加标回收,以回收率考察测定结果的准确度。由于无数据获得磷酸三丁酯的汽化膨胀体积和衬管的具体容积,由于缺乏理论上的最佳进样量,所以,通过逐渐加大进样量,通过考察峰面积的相对标准偏差和回收率,选择最佳进样量。实验结果见表2.3,随着进样量的增加,峰面积逐渐增加,但进样量增加到2.3μL时,增加进样量,但峰面积基本没变;从回收率试验数据来看,进样量为2.3μL体积的回收率反而最低,可以看出此时样品的汽化体积已经超过衬管的容积;进样量为1.8μL时,实验结果精密度和准确度相对较好。
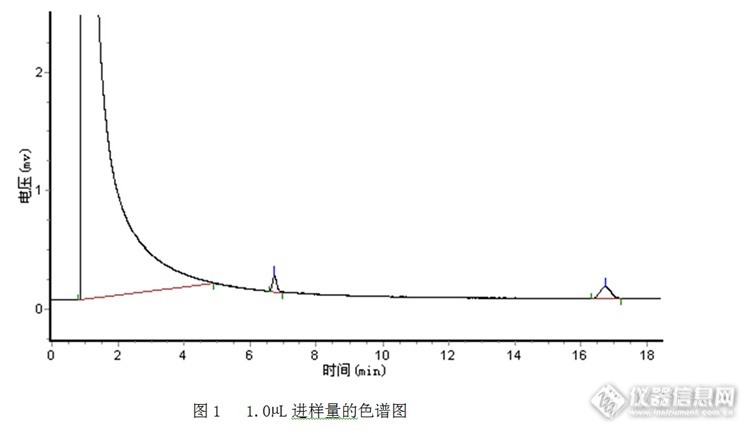
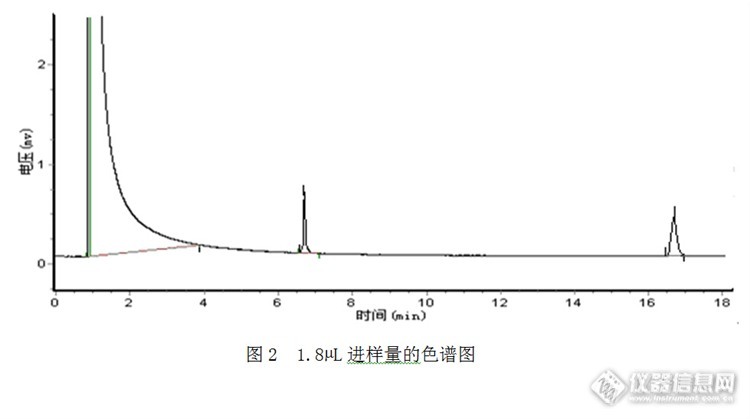
表 样品的进样量与峰面积相对标准偏差、回收率的关系
进样量(μL) | 1.0 | 1.2 | 1.5 | 1.8 | 2.0 | 2.3 |
平均峰面积/mv.s | 2570.4 | 2589.3 | 2610.9 | 2629.8 | 2559.6 | 2346.3 |
峰面积相对标准偏差/% | 4.9 | 4.5 | 3.9 | 2.7 | 1.9 | 0.7 |
平均回收率/% | 95.2 | 95.9 | 96.7 | 97.4 | 94.8 | 86.9 |



































