

雪妖
第4楼2011/08/01
首先恭喜东风恶同志的大作在建军节的第一时间出台了,恭喜恭喜,有几个问题想要请教一下
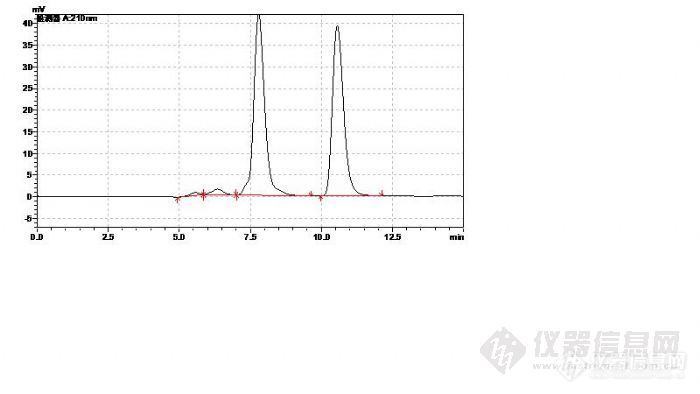
1、第一个手性异构体的样品,您柱子平衡的时候用了多少时间?这个方法是10版药典中的原方法吗?
2、您是要做方法的申报吗?方法验证的时候需要具体做些什么工作?通过药检所审核的几率有多大?因为各个地方药检所的人员对标准方法修改后的接受和理解都有很大的差异?
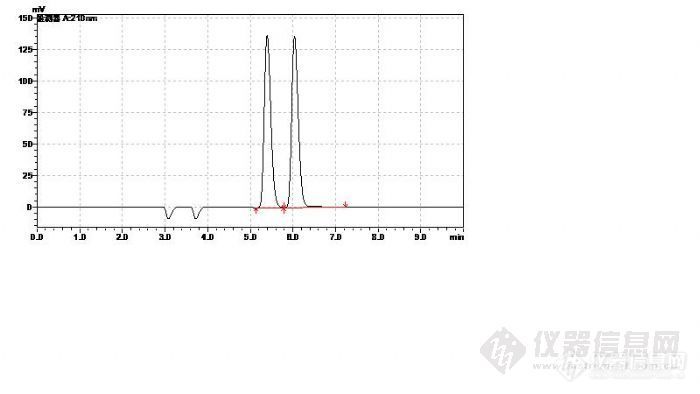
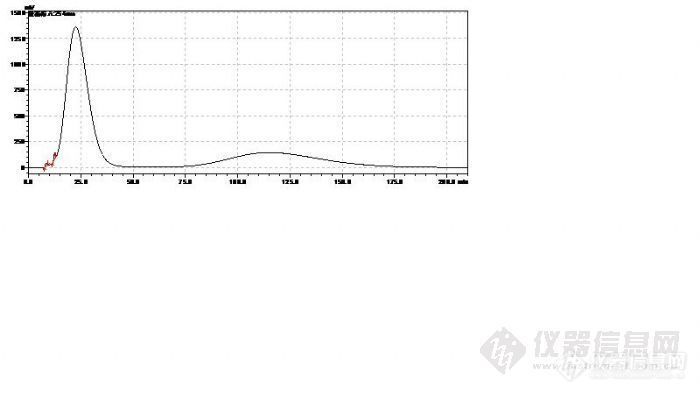
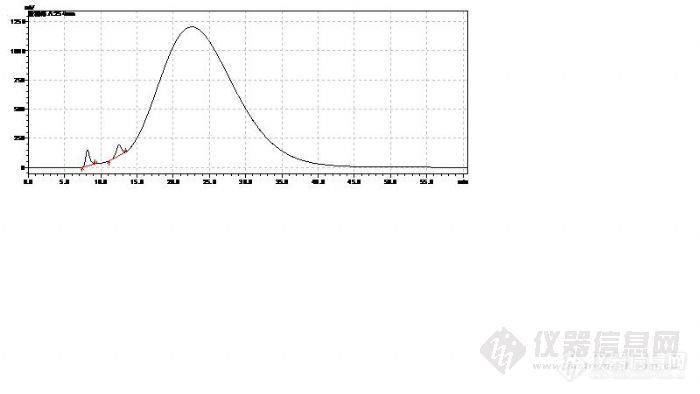
3、凝胶柱后面再加C18,保留时间相差不大,是因为开始的时候是两根凝胶柱,后来用一根凝胶柱 + 一根C18是吧,但是凝胶柱和C18柱的分离机制是不一样的,这样检测聚合物也行吗?
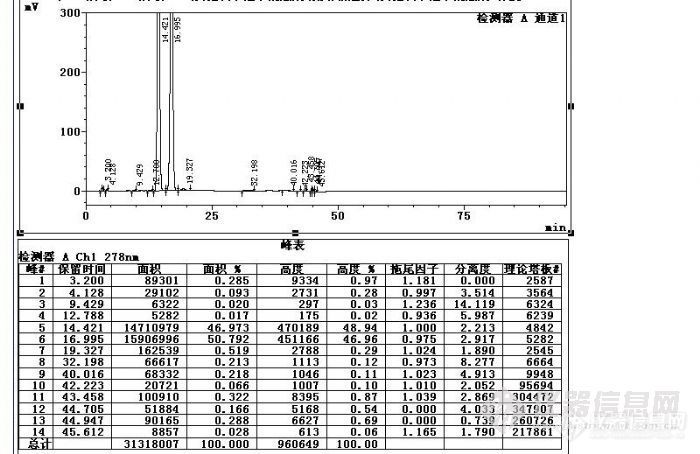
您的样品图中保留时间是9.4min和12.7min这两个是上面标准品中的两个峰吗?那9.4min左右这个是葡聚糖2000了。
这篇文章对我真的有很大的启示,有很多东西也要更深入的学习,先就这几个问题请教一下,谢谢。
东风恶
第6楼2011/08/01
感谢您的关注,谢谢!


































