农残能力验证经验分享----历数近十年的农残能力验证
鹤壁市农产品检验检测中心 张艳丽
能力验证是通过实验室间的比对来确定实验室检测能力的活动,定期对实验室开展能力验证的室间比对,是计量认证和国家实验室认可的特定要求,每年农业部或省农业厅都会对农产品实验室进行能力验证,通过能力验证,可使实验室了解分析项目的整体水平和自己所处的位置,有利于找差距,进一步提高自身技术能力和管理水平。
我从2008年开始参加农残能力验证,一路走来,有欢喜有忧愁,有经验有教训,梳理了一下,分享给大家,希望对大家有所帮助。
一、样品发放模式
1.标液:2008年与2020年发放能力验证样品时,发放了标液,标液浓度为2.0ug/mL,其它年不发标液,让自己配制标液。
2.样品数量:一般为一个空白样品,两个平行样品,但2018年发放时,只发一个空白样品,一个样品,这个样品要做平行样,要从这个样品中称取两回样,进行平行样品测定。2008年与2020年一样,有4个样品,其中1个空白样品,3个平行样品。

图1:发放的能力验证样品
二、考试项目及含量
能力验证的项目逐年增加,从2008年10种农残到现在的90多种农残,从单一标准NY761到今年8个检测标准,仪器从单一气相色谱仪到气相、液相、气质联用、液质联用等。可以说难度也在逐年增加,能够使用不同仪器来完成,也成了对实验人员操作技能的考试。

图2:能力验证项目及检测方法
1. 1.考试时间:
能力验证时间大多集中年初,年中、国庆左右,每年2-3月份出现4次,6月份出现3次,9-10月份出现2次。因此每年过了年,大家都要早做准备,能力验证要来了。
2. 2.考试项目:
从表2来看,农残的考试项目有机磷是甲胺磷、磷胺、甲拌磷、甲基对硫磷、二嗪磷、马拉硫磷、水胺硫磷、杀螟硫磷、甲基异柳磷、伏杀硫磷、甲拌磷亚砜、三唑磷、敌敌畏、毒死蜱等,其中杀螟硫磷3次,甲胺磷2 次,二嗪磷2 次,甲基对硫磷2 次,三唑磷2次,毒死蜱2次,其余均为1次;有机氯及菊酯类多是甲氰菊酯、氰戊菊酯、三氟氯氰菊酯、氯氰菊酯、氟氰戊菊酯、异菌脲、联苯菊酯、氟氯氰菊酯、α-666、γ-666、溴氰菊酯、三唑酮等。其中三氟氯氰菊酯4次,异菌脲3次,氰戊菊酯2次,甲氰菊酯2次,其余1次等。
3. 3.含量:
最低含量是0.042,最高点是0.45,0.10与 0.20出现的次数最多,分别为11次与10次,0.14-0.16出现9次,0.30-0.40出现4次,0.050出现3次。低含量的项目是666,因为它响应值很高,添加量很小,出峰就很好。高含量的是甲拌磷亚砜、治螟磷等,可能与响应值不高有关系。
三、能力验证过程出现的问题及解决方法
1.标液问题
标准溶液,指的是具有准确已知浓度的试剂溶液,常用来做标准溶液绘制工作曲线。在能力验证中标液出现问题比较多,有些是在过程中出现,有些在准备过程中出现,常见的有这几种情况:
1.1标液不出峰
(1)问题
2008年9月27日,做15号样品,省发放标液,标液浓度2.0ug/mL,稀释成0.2ug/mL,发现久效磷不出峰,其它峰也较以前峰面积减小,判断可能有漏气的地方,久效磷的响应度低,在其它峰峰面积减小的情况小,它就不出峰。
(2)解决
检查进样垫、衬管上的O型圈、检查色谱柱两端的石墨垫,发现接检测器的石墨垫有点松,重新拧紧,进样后久效磷出峰。

图3:久效磷出峰情况
1.2标液重合
(1)有机磷标液重合问题
2012年2月21日,做10号样品,自配标液,发现二嗪磷与氧化乐果完全重合,用的是DB1701色谱柱,30m*0.25mm*0.25um,程序升温:温度:80℃保持1min,以20℃速度上升到 130℃,再以5℃上升到200℃,再以15℃上升到250℃,保持11min。进样体积:1uL;不分流进样;检测器300℃;进样口温度:250℃;氮气流速:30mL/min;柱流速:2mL/min;空气流速: 17mL/min;氢气流速14mL/min。
(2)解决
因为10号样品中出峰时间与标液中二嗪磷或氧化乐果重合,所以要判断到底是二嗪磷还是氧化乐果,当时没有气质联用仪,只好在程序升温上想办法,改变程序升温不行,就更换了同型号的VF1701柱,这款柱子出峰较慢,分离效果会好些,在这根柱子上二嗪磷与氧化乐果分离,这样判断出10号样品中添加的是二嗪磷。
图4:标液分离
(3)有机氯标液重合
2016年6月29日联苯菊酯与甲氰菊酯完全重合,用DB-1(30m*0.25mm*0.25um)色谱柱,它是非极性柱子,这两种农药相差时间很少,容易重合,这种情况下只能更换弱极性柱子。
(4)解决
更换DB-5(30m*0.25mm*0.25um)色谱柱,联苯菊酯与甲氰菊酯分开,保留时间相差0.20Min。

图5:标液分离
1.3标液定性
标液一定要用未失效的新打开的,如果标液稀释后放的时间较长,可能会降解,浓度不准,甚至出峰的个数也会不同,准备去拿能力验证样品时,现打开标液,稀释后进样,确定保留时间与峰面积。气相色谱仪以保留时间来定性,如果出现峰重合,定性成了问题。
(1)丙溴磷出几个峰
2020年10月8日,115号样品,准备过程中先进标液确定保留时间,丙溴磷是半年前打开的,进气相色谱仪后,出现两个峰,但是用新买的丙溴磷打开后,只出一个峰。
(2)解决
将新买的两支丙溴磷都打开,进行稀释进样,只出一个峰,同时盲样样品中也只出一个峰,确定旧丙溴磷降解,不能使用。标液最好现用现买,放的时间长了会降解。
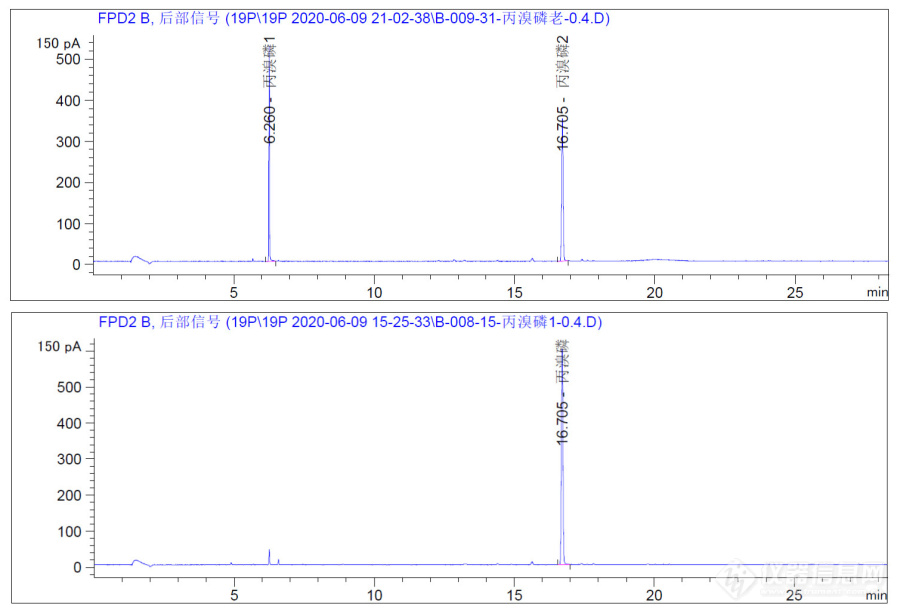
图6:丙溴磷出峰情况
(3)三唑酮、三氯杀螨醇的定性
气相色谱仪以保留时间定性,当两个农药完全重合时,无法判断,这时要更换柱子或用气质联用仪或液质联用仪进行判断。2020年的能力验证时,三唑酮、三氯杀螨醇完全重合。
(4)解决
如果想让它们分离,要换不同极性的色谱柱,比如要用1701柱,还可以用气质联用仪,用MRM方法扫描,用4个离子对来判断。

图7:气质联用仪定性
1.4标液重现性
上仪器检测时,上机顺序是这样的,要先进标液,然后进空白样品,再进盲样样品,如果盲样又做了第二次,我们还会按这个顺序再上机,这时如果标液重现性不好,就无法计算样品的结果。
(1)峰面积越来越小
2012年2月21日,做10号能力验证样品,配甲拌磷与二嗪农混标0.2ug/mL,第一天早上进样、晚上进样、第二天早上进样,甲拌磷标液的峰面积越来越小,但二嗪农的峰面积稳定。
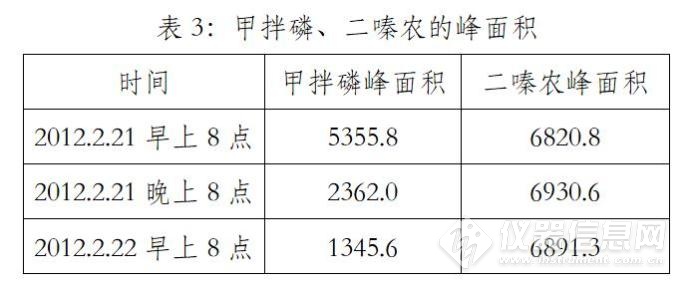
(2)解决
配混标进样,如果两种标液峰面积同时增大或减小,与仪器有关,可能与进样垫、衬管、色谱柱等漏气或污染有关,但一种标液正常,另外一种标液峰面积不断减小,可能与标液质量有关,我们打开另外一支新的甲拌磷,还是同样的问题,可能是这一批次的标液都有问题。于是借了其它实验室的一支甲拌磷,稀释后进样,两天内峰面积稳定。
(3)峰面积越来越大
配制敌敌畏、甲基对硫磷的标液0.5ug/mL,随着进样次数增加,峰面积逐渐增大。
(4)解决
峰面积逐渐增大,与衬管污染、色谱柱污染有关,色谱柱要老化,切柱头,需要关机再开机,时间太长。所以可以先换衬管,用新的高惰性衬管,再次进样,峰面积稳定。
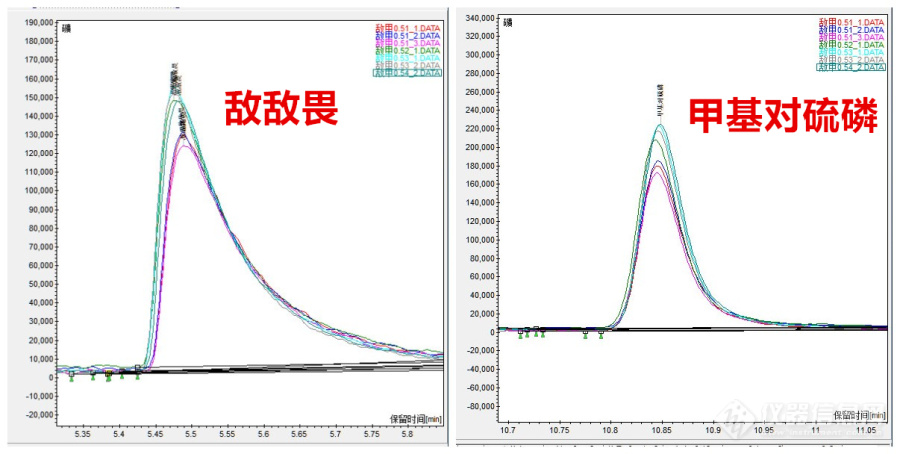
图8:峰面积越来越大
1.5标液配制计算错误
(1)标液配制问题
标液要稀释后才能进样,一般打开标液100ug/mL,稀释成10ug/mL,然后稀释成0.2ug/mL,进气相色谱仪,发现标液不出峰。查找仪器条件,一切正常。
(2)解决
重新配制标液,进气相色谱仪,出峰正常,原来是太紧张,计算时错误,吸的标液量少10倍,导致标液含量0.02ug/mL,含量太低,有些农药不出峰。计算时结果是毫升,要换算成微升,用吸液枪来完成,很容易在换算时出错。
2.前处理问题
日常工作中要加强对前处理的练习,对于平行样品不平行、回收率低等问题,查找原因并解决,这样才能在能力验证时,一次成功,保证数据的准确性。
2.1平行样品不平行
(1)前处理问题
2012年的能力验证,10号样品,前处理是NY761-2008,两个平行样品的结果相差近一半,相对偏差较大。
(2)解决
先排查了仪器的问题,认为问题出在前处理上,取空白样品,在样品中做加标试验,氮吹时在同一根氮吹针上完成,保证两个平行样品氮吹近干的状况一致,两个平行样品结果相近,取盲样所剩的乙腈液体,在这根氮吹针上重新做前处理,得出的数据与原样品平行,原来是不同的氮吹针上调节的气流不同,最后氮吹近干的状况没有把握好,导致平行样品不平行。
(3)样品问题
2018年能力验证时,发了一盒笋瓜、一盒苹果样品,让从每盒里取3个平行样品,进行检测,得出的结果不平行。
(4)解决
将盒里样品进行超声、匀浆,进行混匀后,再次取样进行检测,平行样品较平行,但因为样品里已经取过样,数据的准确性受到影响。
当寄送的能力验证样品是一个空白样和一个阳性样,需要参加实验室自己称量样品的情况时,这时候需要一次性解冻所有样品,用玻璃棒充分搅匀,再进行超声,混匀器上混匀后,把所有阳性样同时分装成三等份样品。
取一份先进行检测,也就是进行预实验,即定性实验和初步定量实验,需要通过对比加标样与空白样确认出添加的农药和它们各自的添加水平,等到预实验结果确定后,再拿出另外两个加标样做正式实验,同时进行与加标浓度同一水平的加标实验,计算不同农药的回收率,这样处理是为了避免样品多次解冻、多次称量造成的误差。
2.2回收率低
能力验证时,除了做盲样,同时要做加标回收,确保自己的前处理过程及仪器没问题,如果加标回收率低,就要查找原因了。
(1)问题
甲胺磷、敌敌畏、氯氰菊酯、氟氰戊菊酯等都出现过回收率低的问题。有机磷回收率低大都出现在氮吹环节,有机氯及菊酯类大都出现在氮吹、过弗罗里析柱环节。
(2)解决
有机磷类在氮吹时,氮吹近干要掌握好,里面有一滴时就要拿出来,用吹耳球吹一下,底下潮湿即可。如果烧杯底下完全干了,回收率就低了。
有机氯及菊酯类在氮吹时同样要注意这一点,同时在过弗罗里析柱,要先淋洗小柱,再倒入样品液,一定要注意小柱不能干。洗脱过程要自然流下,不要用外力挤压,容易造成平行样品不平行。

图9:氮吹、过弗罗里析柱
2.3空白样品含目标物
(1)问题
2013年能力验证时,空白中含有百菌清,但平行样品中不含百菌清,这个数据报不报?如果平行样品中也含百菌清呢?
(2)解决
当空白中含有某种农药时,要与能力验证发放单位沟通,一般会让上报,如果空白中有,盲样样品中也含有,也要与发放单位沟通,一般会扣除空白中的含量再上报数据。
3.仪器问题
仪器在能力验证时很重要,有很多问题出在仪器上,所以准备工作要保证仪器稳定,进样口干净等。
3.1基质效应
农残检测基质效应是绕不过去的坎,要消除基质效应要用基质配制标液,用高惰性衬管减少目标物的吸附。
(1)问题
2014年能力验证时,做14号样品,用丙酮配制混标,同时做加标回收,发现水胺硫磷的回收率高达140%,其它农药的回收率也较高。
(2)解决
用笋瓜基质配混合标液,用高惰性衬管进样,这几种农药的加标回收率正常,然后用这个标液的峰面积来计算盲样,得出最终结果。
3.2重现性不好
(1)问题
重现性不好,多是漏气、衬管污染、色谱柱污染、进样针堵塞等引起的。2020年6月8日能力验证时,样品峰面积,连进两针,它的重现性相差21%。
(2)解决
我们检查了进样垫、衬管、色谱柱两端的石墨垫等,都没发现问题,最后查到进样针,进样针无折断,卸下清洗时,发现针芯有一段变黑,可能是磨损所致,更换新的进样针,重新进两针,重现性在3%以内,问题解决。
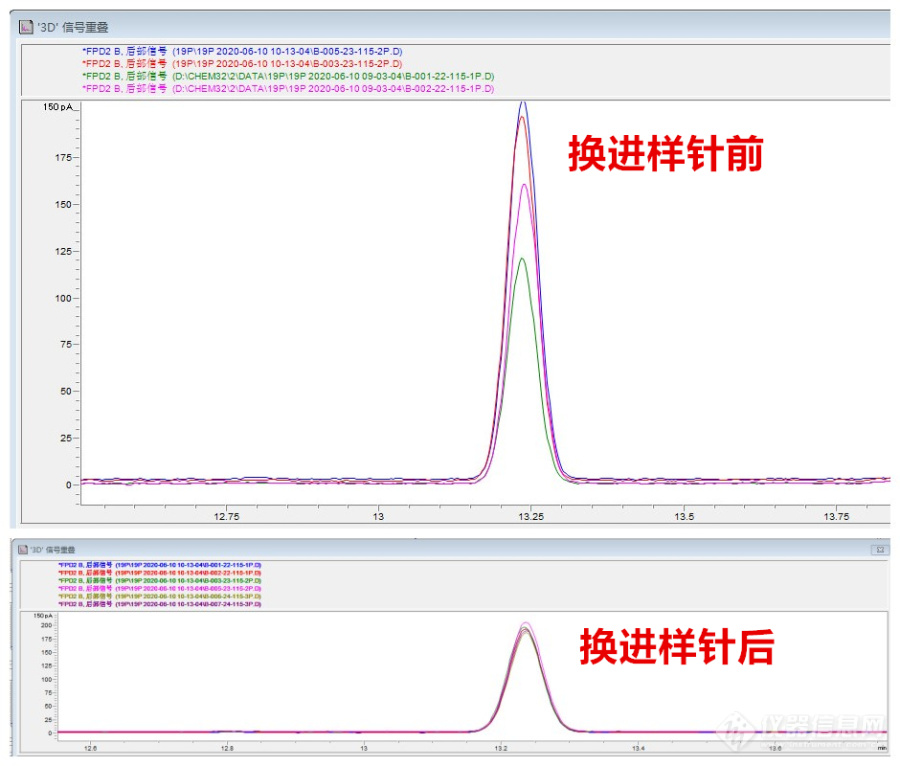
图10:进样针引起的重现性不好
3.3出平头峰
当样品中目标物出现平头峰时,如何解决?
(1)问题
2020年6月8日能力验证时,三唑酮标液配制0.1ug/mL时出现平头峰,盲样进样后,三唑酮也出现平头峰。
(2)解决
出现平头峰,说明浓度超载,峰面积就不准确了,我们配制了0.1ug/mL与0.2ug/mL的三唑酮,浓度相差一半,但峰面积变化不大,说明平头峰的峰面积不会再增加多少。解决平头峰有两种方法:
一是稀释样品,将样品再加溶剂进行稀释,标液也同样进行稀释,稀释时要注意计算准确,得出准确的稀释倍数。
二是调节仪器的灵敏度,我们用的是天美气相GC450,ECD检测器有1与10两种灵敏度,数字越大,响应值越大,峰面积越大,两者相差10倍,调节成1后,重新进样,不再出平头峰,峰高且尖锐,由此计算出样品含量。
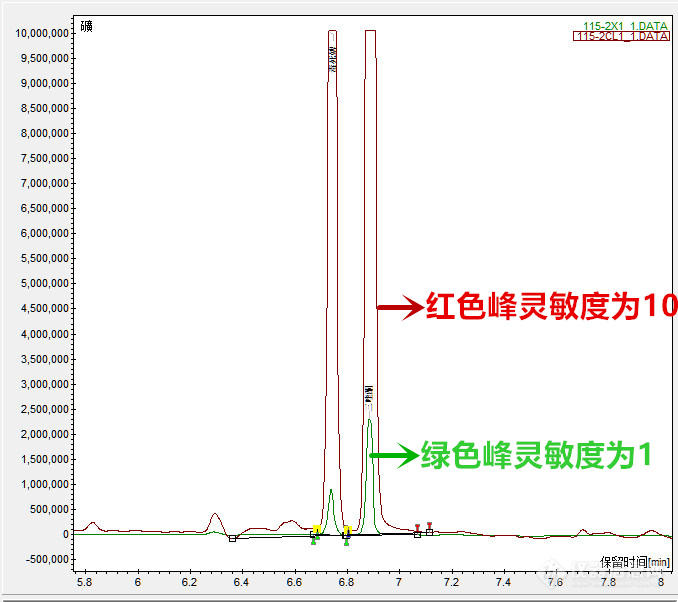
图11:调节灵敏度
四、能力验证的经验分享
评价参加实验室的检测能力,通常用Z比分数进行评价。
Z值的绝对值小于等于2,满意、通过;
Z值的绝对值大于等于3,不满意、不通过;
Z值的绝对值小于3大于2,可疑。

随着能力验证的项目与难度逐年增加,做能力验证时以下几点要注意:
1.仪器
1.1色谱柱
要特意准备一支新的色谱柱,只做能力验证,每次能力验证都用它,用了几次后,转成检测任务的柱子,这样不浪费。因为柱子是干净的,所以不易出现污染后的各种不正常数据。
1.2进样口
进样口要保持干净,能力验证时,用新的进样垫,用新的衬管与新的O型圈,色谱柱上的石墨垫也换成新的,进样针用新的,或检查进样针没问题再使用。这样避免了重现性不好、回收率高问题。
2.检测标准的前处理
2.1氮吹或旋蒸
样品浓缩时,目标物容易损失,也容易造成平行样品不平行,氮吹时要保持气流量一致,但是氮吹仪上没法检测流量,只能用笨办法,将上面的气流旋钮都放到最大,在不同氮吹针头下进行氮吹,记录氮吹完的时间,时间相近的画上记号,在做平行样品时,放在有记号的氮吹针下,可以保持平行性。
旋转蒸发时,转速要由慢到快,保持转速200左右,水滴成滴滴下,不可成线,过快时要关掉真空泵。旋转蒸发近干要严格控制好,蒸发太快、蒸发温度太高都会导致回收率降低。
2.2过小柱
净化时淋洗弗罗里析柱时,液面不要干,当溶剂液面到达柱吸附层表面时,立即倒入上述待净化溶液,如果柱子表面干了再倒入溶液,农药会吸附在柱子上,回收率降低。
净化时,淋洗液与洗脱液要按标准上规定的顺序进行,如果溶剂用错,或顺序弄错,目标物在小柱上洗脱不下来,回收率会很低。
2.3混匀、定容
用丙酮清洗小烧杯的待测液时,最好在混合器上转圈洗烧杯底部,移入15mL刻度离心管中,用约3mL丙酮分三次洗烧杯,洗烧杯时也在混合器混匀清洗。
用刻度离心管定容时,刻度离心管要用检定过的,定容时要准确,定容后要再次混匀。

图12:前处理图片
3.基质效应
大多数农药的回收率会在70-120%之间,但是甲胺磷、氧化乐果等强极性农药,有较强的基质效应,回收率会高达400%,解决的办法是同一样品的基质配标液,例如找一空白样品,按方法进行前处理,最后定容至5mL,用这5mL配制标液,进气相色谱仪测得峰面积,用来计算样品中甲胺磷、氧化乐果的回收率,这样就大大减少了基质效应。
3.1标准曲线
在农残实验中,不同基质对不同农药的基质效应都是不一样的,所以在最终结果的校正时一定要配置基质曲线,因为能力验证给的样品量很少,得到基质很少,可以先与样品发放单位沟通,询问用的是什么样品,然后购买该蔬菜样品,清洗干净,进仪器确定没有农药,然后进行前处理,得到基质,配标准曲线,用不完的基质可以冷冻保存。
3.2单点校正
标曲做好后,先计算盲样结果,得出数据后,再用基质配与盲样结果相近的单点标液,再进行精确计算,得出准确数据。
也可以不配标准曲线,用基质先配单点,计算出盲样结果后,再用基质配相近的浓度。
4.加标回收
加标回收率低要从前处理方面找原因,大都集中在前处理方面,从提取、浓缩、净化、定容等方面找原因,提取时用刻度吸管吸液要准确,浓缩时氮吹或旋蒸,近干状况要掌握好,净化时过小柱,小柱不能干,淋洗或洗脱溶剂顺序不要错,定容时要准确,要注意混匀。
加标回收率高要从基质效应来查找问题,基质配标液,用高惰性衬管、称取样品要混匀等,称取样品时,一定要混匀样品后再称样,我们遇见过称取的菜汁多,称取菜样少,引起回收率高的情况。
5.加强培训与练习
为了顺利通过能力验证,要加强培训与练习,让有经验的同志进行培训,对能力验证出现的问题进行总结,对关键点进行详细介绍,组织要参加能力验证的人员进行日常练习,进行加标回收等试验,对出现的问题及时解决,最好对每种农药都要做加标回收试验,记录它们的回收率,对回收率低的与高的农药,要查找原因,找到解决办法。

图13:培训与练习
五、能力验证总结
说几点大家关心的问题,是本人的一点总结,不对的地方请指正。
1.要不要与其它人对答案
每次能力验证时,大家都会建个群,在里面热闹的对答案,因为参加能力验证的机构比较多,一家一个添加也是不现实的。大都分是分组进行添加的,这样肯定有人与自己的含量是一样的,有时还真能找对人,然后大家对答案,答案一样心里有底。不过我不建议这样做,一是茫茫人海,寻找与自己一组的人,很费时间,二是对方的答案可能是错的,本来自己做对的,因为不自信,反而不通过。
2.要不要用回收率倒推结果
这个问题也经常有人问,我们在做能力验证时,同时会做加标回收,看它的回收率如何,如果加标样品回收率在80%-120%之间,就直接报结果,如果加标样品的回收率低于70%,要查找前处理的问题,如果时间很紧,可以倒推一下,但不能保通过,有时加标样品做的回收率低了,可盲样做的没问题,一倒推,反而数据不对了。如果回收率高了,那就换个衬管或用基质配标液,消除一下,再计算结果。
我们的成功经验是,前期准备工作充分,在能力验证前将每种农药的加标回收都做一遍,并做好记录,对于加标回收低或高的,一定要找到解决方法,这样手中有数,心里不慌,不管考试哪个,都有应对措施。
3.如果不平行,怎么办?
一是平常要多练习平行样品的测定,比如每种农药都加标平行样品的测定,对不平行的原因要找到。二是如果盲样真出现严重不平行,相差一半也是有的,那么就报高数据的,低数据的一般都通不过。比如平行一0.05,平行二0.10,那就报0.10,不过不平行的原因接下来一定要找出来,如果找不出来,下次能力验证还是这个问题。
4.多仪器互做验证
做能力验证时,可以利用不同仪器互做验证,比如用气相色谱仪检测农残时,判断是否该农药,就可以用气质联用来定性,有些在气相色谱仪、气质联用仪上容易吸附的农药,可以用液相色谱仪、液质联用仪来检测,但是要用该仪器的前处理方法,这样数据才能准确些,比如用NY761做前处理,应该上气相色谱仪来检测,如果拿到液质联用仪上,也可以出数据,但数据的准确性还是以气相色谱仪为准。
回首这近十年的能力验证,一路坎坷,一把辛酸,没少通宵达旦做试验,没少为报哪个数据而发愁,没少查找各种问题,累得两鬓生白发,最终也练就了一身过硬本领。能力验证年年在路上,祝愿大家都能顺利通过能力验证!



































